No se publica un libro sin alguna divergencia entre cada uno de los ejemplares. Los escribas prestan juramento secreto de omitir, de interpolar, de variar. [No book is published without some divergence between each of the copies. Scribes take a secret oath to omit, to interpolate, to change.] (Jorge Luis Borges, La lotería en Babilonia, in Ficciones, 1962)
This is the first on series of posts on stemmatics, a field just as much in love with trees and networks as are phylogenetics and historical linguistics. Being an introduction, I explain what the field does, present the most important jargon, and offer a list references that, while suitable for the audience of this blog, is denser than what one might expect for a blog post.
Thank you to Mattis and David for inviting me to write!
Textual criticismTextual criticism (or, less precisely, "philology") is a discipline concerned with the investigation of the history of literary, legal, and religious texts for explaining how differences among the copies of a text (its "witnesses") arose, and with the production of "critical editions", either scholarly curated versions of a text that aim to reconstruct the lost original or corrected versions of an existing copy.
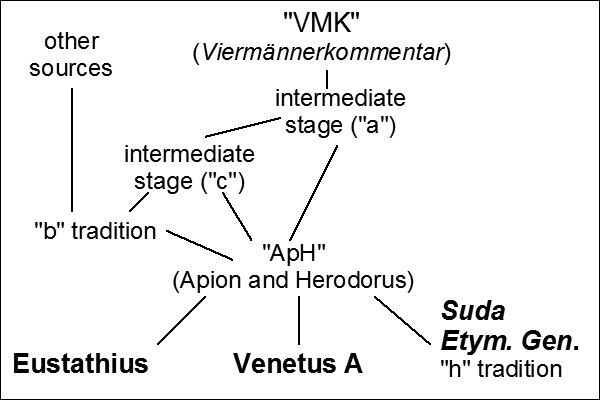
The problem of divergence between copies of text, with the accumulation of involuntary and deliberate errors, as well as the need for a systematic study of such differences, is as old as writing itself. For example, our current editions for the epic poems of Homer descend from Ancient philological attempts to restore an uncontaminated original (see the first two figures). These include the edition of Pisistratus (VI century BCE, which determined what was to be sung at the Panathenaic Games), and the so-called VMK (
Viermännerkommentar, "commentary of the four men") of the Alexandrian School (I-II century BCE), which is generally assumed to be the root of the witnesses that we have.
 |
Van der Valk's reconstruction of the sources for Venetus A, one of the most
important manuscripts of Homer's Iliad (source: Wikipedia). |
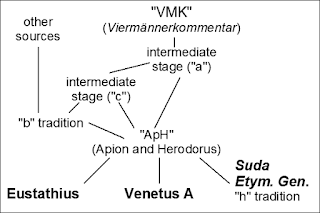 |
Erbse's reconstruction of the sources for Venetus A, one of the most important
manuscripts of the Iliad (source: Wikipedia). |
Before stemmatics, an edition could either be based on a "good copy" (a version considered to be less contaminated or more faithful than others), in a "majority reading" (in which the most attested variant would be chosen), or in a principle of "eclecticism" (with each best reading individually selected by the editor's judgment). Each new version, as expected, contributed even more to the confusion, particularly when changes were voluntary.
Among the texts with long and complex traditions, objects of countless and sometimes bloody disputations on the "correct" readings, are the Bible and codes of laws, for which it was not uncommon to have a different version in each city, with predictable consequences. For example, the first published textual tree, as already covered in this blog (
The first Darwinian evolutionary tree), was authored by Carl Johan Schlyter in 1827 in a study precisely on the multiple and conflicting copies of Swedish law.
As such, it is no surprise that objective approaches were soon developed (Homer's VMK edition being one of the first examples), culminating with the development of stemmatics, with its study of the genealogical relationship between witnesses, and its representation of such relationships by means of trees.
StemmaticsAs a scientific approach to textual criticism, stemmatics established itself from the beginnings of 19th century as an alternative to emendations based in the opinions and wishes of editors, possibly inspiring both Charles Darwin and August Schleicher (for a general discussion on the development and significance of this method, see Timpanaro 2005). However, more than a "source", we should consider it a branch equally stemming from the "cultural framework" (Macé and Baret 2006: 91) that also gave us Darwinism and historical linguistics.
As was true for these latter disciplines, stemmatics was at first opposed, because of the revolution it brought to its field, along with its genealogical trees. However, just as in these sister disciplines, the results of the new mindset introduced by the explanation of evolution with trees could not be ignored, and this approach is so central to textual criticism that the latter can be divided into periods before and after the work of Karl Lachmann, the "father" of stemmatics, in particular the publication of his edition of Lucretius'
De rerum natura (1850). In his commentaries, besides demonstrating the number of lines per page in the lost manuscript at the root of the tradition, Lachmann was even able to demonstrate the kind of script used to write it (Lachmanni 1850).
The work he chose, with the importance of Lucretius in the development of the scientific mindset (and, as we should remember when dealing with cultural evolution, of Darwin's theories), is unlikely to be casual, but this is a matter for a different blog post.
TreesGenealogical trees are so central to the stemmatic method that the field itself is actually named after them. The main goal of an editor is to produce a
stemma codicum ("family tree of manuscripts"), or simply
stemma, a tree-like structure that supports the textual emendation and represents the "tradition" (the witnesses' genealogy), in analogy with the family trees of Roman families that figured in many texts reviewed by 19th century philologists. Stemma, in fact, is a Greek word meaning garland or wreath, that was incorporated in Imperial Latin to designate a family tree (and, figuratively, nobility itself), as family trees were drawn with a stemma at their top.
In short, stemmatics begins with a
recensio, which is an investigation of all total and partial copies of a work. This review is followed by a
collatio, a systematic scrutiny of the manuscripts' contents, when readings are aligned and compared. The results of this alignment are used to produce the
stemma, following the principle that "community of errors implies community of origin". By analyzing the
stemma and the errors, editors finally proceed to the
emendatio, which is a reconstruction that explains the known variants, and is intended to represent the "archetype" (a lost witness at the root of the ramification, assumed to be closer to the original than any other copy).
A
stemma is conventionally drawn top-to-bottom, with vertical placements roughly indicating the date of the manuscript (the higher, the older). Solid edges ("arrows") indicate descent, while dashed ones imply contamination (scribes using more than one source). Witnesses are usually labeled with abbreviated names or Latin letters, when the manuscript is available, or with Greek letters, when it is missing (with α usually reserved for the archetype and ω for the original). Below is a reproduction of Petrocchi's partial stemma for the tradition of Dante Alighieri's
Divine Comedy, which I will cover in a future post. Note that the genealogy is actually a reticulating network rather than a simple tree.
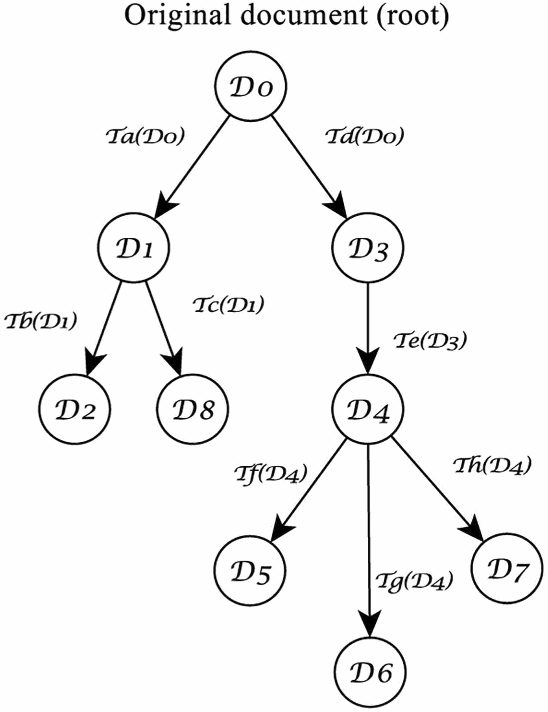
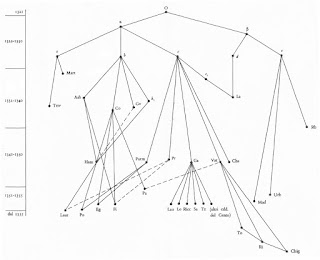 |
Petrocchi's partial stemma for the Divine Comedy, presented in the
introduction to his critical edition (1965). |
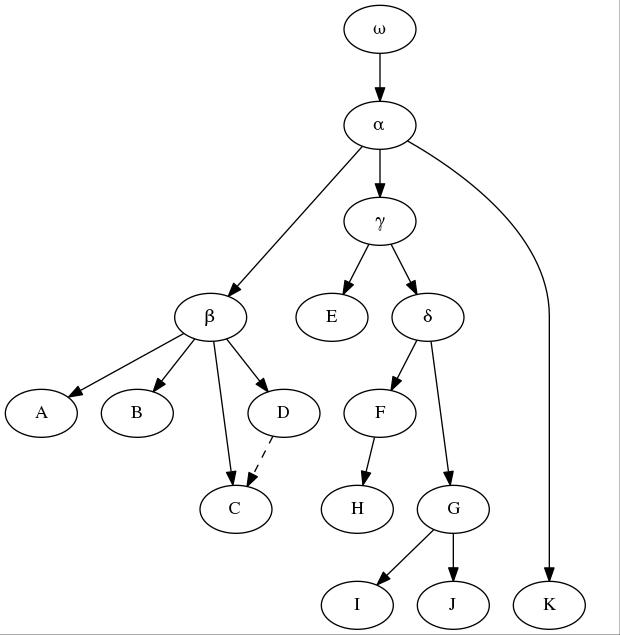
The example
stemma offered by Maas (1958), adapted below, is still useful to demonstrate the principles of stemmatics. In this example, for a textual emendation manuscript H should be eliminated (as it descends from F), as well as I and J (copies of G). Manuscript C shows a contamination from its collateral D, something which should be considered when weighting errors. Sub-archetypes β and γ are to be inferred from the available witnesses of their branches, and their readings will have the same weight as K, the only member of the third family branching from the archetype (even though it is a recent manuscript), in establishing the "lesson" of α. Errors might be presumed in α itself, or even in the original ω, and in both cases a corrected "lesson" might be offered by the editor after internal and external evidences.
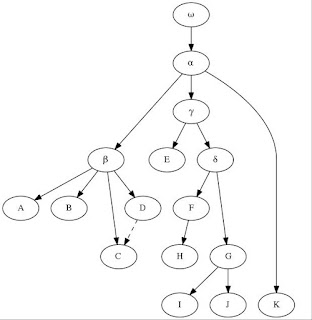 |
| Exemplary stemma adapted from Maas (1958). |
Adoption and practice Stemmatics has been criticized and confronted since Lachmann's time. It requires very specialized knowledge, for example in distinguishing between monogenetic and polygenetic errors, i.e. those that arose once and those that emerged independently more than once (and that, as such, are not disjunctive). A number of its suppositions are routinely called into question, such as the idea that each copy always derives from a single source (accepting contamination, at most), that each copy has at least the same number of errors of its source, and, fundamentally, that traditions have one and only one archetype.
Many measures tend to be adopted to reduce the editorial effort. These include eliminating manuscripts considered to be
descripti (i.e. proved to descend from a preserved witness, in theory sharing all the errors of their sources), and only performing the
collatio in a set of critical passages (
loci critici). While a complete
stemma and a full
collatio are desirable, such compromises might be unavoidable for long texts with ample traditions. For example, in the case of Dante Alighieri's
Divine Comedy, after considering the time employed by scholars such as Petrocchi, Sanguineti, and Shaw for their editions, Trovato (2016) estimated the length of a full stemmatic approach in 400 man-years.
An alternative to stemmatic methods and suppositions, which also reduces the editorial effort, is found in scholars who follow the work of Joseph Bédier, who successfully challenged the limits of stemmatics by adopting a renewed version of the method of the "good copy" for his editions of medieval texts. The Bédierian method does not refute a scientific approach or methods such as the
recensio, the
collatio, or even the production of a stemma, but these are used to support the editor's judgment in selecting and curating a
bon manuscript — a good edition of text to be corrected only where errors can be proved beyond reasonable doubt. In short, trees (and networks) have been central to textual criticism even when stemmatics itself, as a method, is being challenged.
Considering the editorial effort and the analogies with linguistics and biology, it is no surprise that digital workflows have been proposed, along with the development of computer resources and phylogenetic methods. Ideas for new approaches were explored by Froger (1969), and formal phylogenetic methods were attempted by Platnick and Cameron (1977). Recently, the number of editions supported by formal phylogenetic methods and software has increased (see, for example, Barbook et al. 1998; Stolz 2003; and Lantin, Baret and Macé 2004), also in the face of scientific evaluations of performance (Roos and Heikkila 2009).
Besides advances in speed and replicability, the new technologies are allowing us to expand the goals of the discipline, moving from electronic editing to computational philology. In fact, while the field has for centuries been defined by the production of critical editions, digital approaches have been shown to support a reduction in the importance of "authorial intention", allowing researchers to focus on the reception of texts by the public, in line with developments of literary theory (Jauss 1982), and with the goals established by the "New Philology" (Cerquiglini 1989). Manuscripts with readings that differ from a supposed original, traditionally described as "corrupted", are changing from copies that were meant to be discarded into data points that collaborate in an investigation of human history that is assisted by quantitative data and methods.
ReferencesBarbrook A.C., Howe C.J., Blake N., Robinson P. (1998) The phylogeny of the Canterbury Tales.
Nature 394 (6696): 839.
Cerquiglini B. (1989)
Éloge de la variante: histoire critique de la philologie. Aux Travaux. Paris: Éditions du Seuil.
Froget J. (1969) La critique des textes et son automatization.
Bulletin De L’Association Guillaume Budé 1(1): 125–129.
Jauss H.-R. (1982)
Toward an Aesthetic of Reception. Minneapolis: University of Minnesota Press.
Lachmann C. (1850)
De Rerum Natura. Commentarius. Berolini: Imprensis Georgii Reimeri.
Lantin A.-C., Baret P.V., Macé C. (2004) Phylogenetic analysis of Gregory of Nazianzus’ Homily 27.
7èmes Journées Internationales d’Analyse statistique des Données Textuelles, pp. 700-707.
Maas P. (1958).
Textual Criticism. Translated by Barbara Flower. Oxford: Oxford University Press.
Macé C.; Baret P.V. (2006) Why phylogenetic methods work: the theory of evolution and textual criticism.
Linguistica Computazionale. The Evolution of Texts: Confronting Stemmatological and Genetical Methods 24: 89–108.
Platnick N.I., Cameron H.D. (1977) Cladistic methods in textual, linguistic, and phylogenetic analysis.
Systematic Zoology 26: 380–385.
Roos T., Heikkilä T. (2009) Evaluating methods for computer-assisted stemmatology using artificial benchmark data sets.
Literary and Linguistic Computing fqp002.
Stolz, M. (2003) New philology and new phylogeny: aspects of a critical electronic edition of Wolfram’s Parzival.
Literary and Linguistic Computing 18(2): 139–150.
Timpanaro S. (2005)
The Genesis of Lachmann's Method. Translated and edited by G. W. Most. Chicago: University of Chicago Press.
Trovato P. (2016)
Metodologia editoriale per la Commedia di Dante Alighieri. Ferrara. See
Youtube; date of access: March 19, 2017.