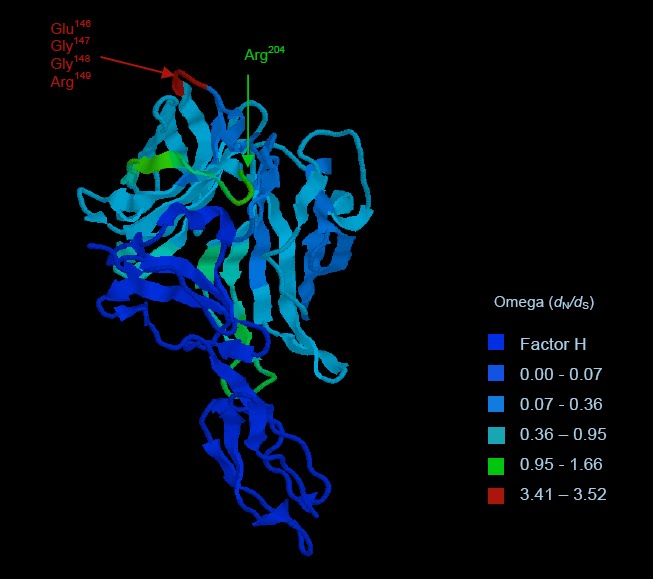
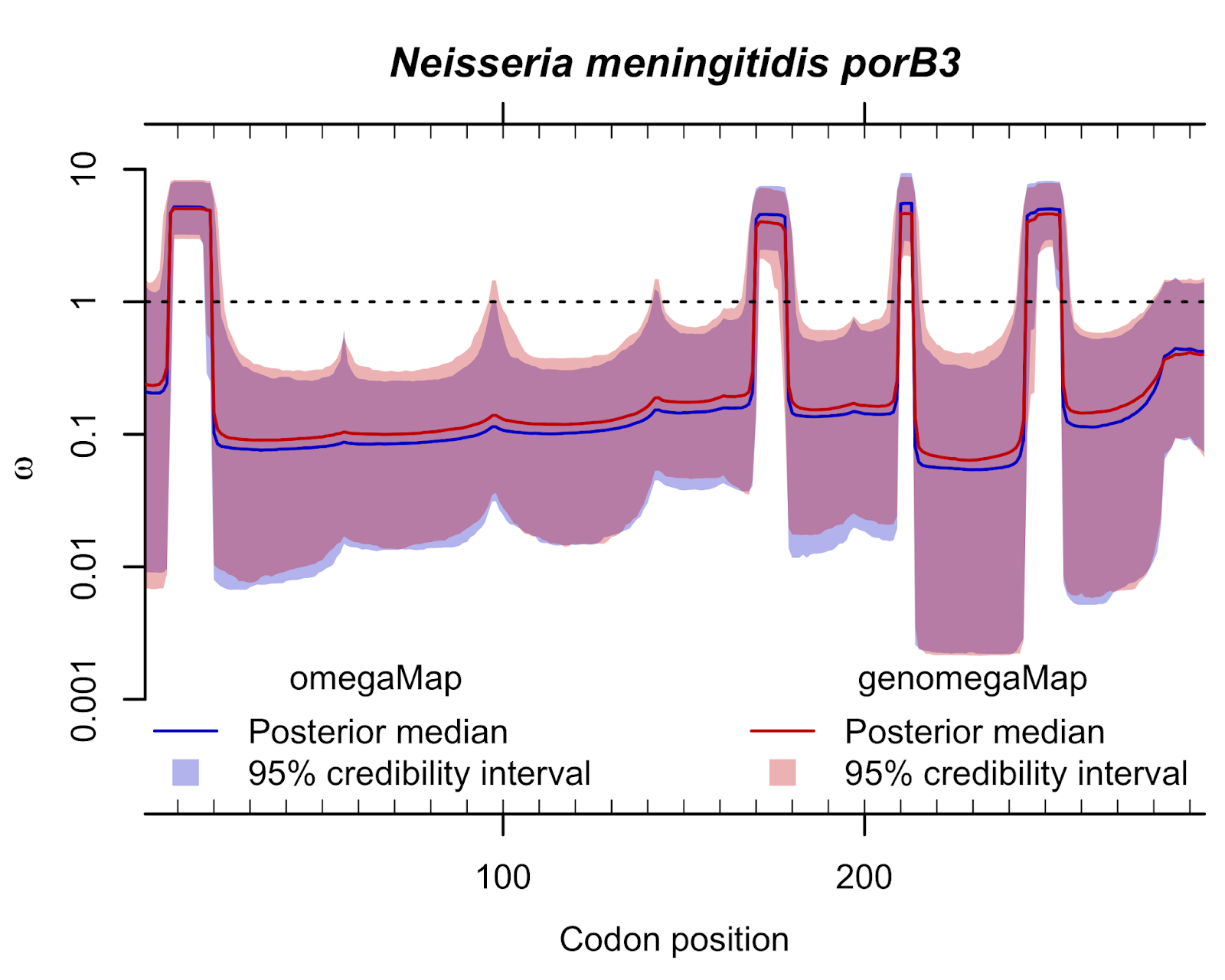
 Published this week in Molecular Biology and Evolution, is a new paper joint with the CRyPTIC Consortium "GenomegaMap: within-species genome-wide dN/dS estimation from over 10,000 genomes".
Published this week in Molecular Biology and Evolution, is a new paper joint with the CRyPTIC Consortium "GenomegaMap: within-species genome-wide dN/dS estimation from over 10,000 genomes".The dN/dS ratio is a popular statistic in evolutionary genetics that quantifies the relative rates of protein-altering and non-protein-altering mutations. The rate is adjusted so that under neutral evolution - i.e. when the survival and reproductive advantage of all variants is the same - it equals 1. Typically, dN/dS is observed to be less than 1 meaning that new mutations tend to be disfavoured, implying they are harmful to survival or reproduction. Occasionally, dN/dS is observed to be greater than 1 meaning that new mutations are favoured, implying they provide some survival or reproductive advantage. The aim of estimating dN/dS is usually to identify mutations that provide an advantage.
Theoreticians are often critical of dN/dS because it is more of a descriptive statistic than a process-driven model of evolution. This overlooks the problem that currently available models make simplifying assumptions such as minimal interference between adjacent mutations within genes. These assumptions are not obviously appropriate in many species, including infectious micro-organisms, that exchange genetic material infrequently.
There are many methods for measuring dN/dS. This new paper overcomes two common problems:
- It is fast no matter how many genomes are analysed together.
- It is robust whether there is frequent genetic exchange (which causes phylogenetic methods to report spurious signals of advantageous mutation) or infrequent genetic exchange.
The paper includes detailed simulations that establish the validity of the approach, and it goes on to demonstrate how genomegaMap can detect advantageous mutations in 10,209 genomes of Mycobacterium tuberculosis, the bacterium that causes tuberculosis. The method reproduces known signals of advantageous mutations that make the bacteria resistant to antibiotics, and it discovers a new signal of advantageous mutations in a cold-shock protein called deaD or csdA.
Software that implements genomegaMap is available on Docker Hub and the source code and documentation are available on Git Hub.
With the steady rise of more and more genome sequences, the analysis of data becomes an increasing challenge even with modern computers, so it is hoped that this new method provides a useful way to exploit the opportunities in such large datasets to gain new insights into evolution.