Following the current flow, we have had a few recent coronavirus posts here on the Genealogical World of Phylogenetic Networks. In this post, I'll show the results of a little experiment coming back to David's original post on the topic. Can we use trees to "fish" for evidence of recombination?
As David pointed out, even when we use a phylogenetic-tree inference method to analyze virus genomes, we don't really end up with a phylogenetic tree. Instead, we have a tree reflecting genetic similarity, which will reflect the phylogeny to some unknown extent. The main problem with virus genomes, however, is that they easily recombine — and thus different parts of a virus genome may have different evolutionary histories. A single tree cannot reflect this.
This does not mean that trees cannot tell is something about virus evolution. However, these trees become part of a fishing exercise, looking for different possible historical pathways, which may reflect recombination events.
The tree
Our SARS harvest matrix includes about a dozen sequence groups, which we have labeled Type 1 (the original SARS-CoV) to 9b. Type 7 is the new SARS-CoV-2. For my experiment here, I picked one place-holder sequence per main type (to speed up calculation time). I added two more types: the newly found direct sister of SARS-CoV-2; and some "unclassified" SARS-like viruses from pangolins, which earlier were proposed as sisters, as shown in this tree from the GISAID web page.
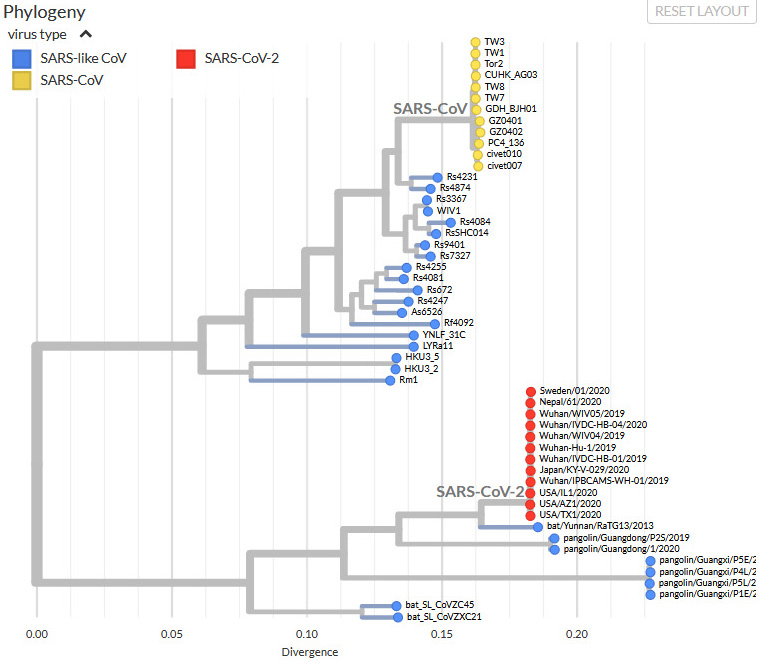 |
| The phylogenetic neighborhood of SARS-CoV-2 (GISAID, screenshot captured 3/6/2020). Note the flatness of the CoV(-1; yellow) and CoV-2 (red) subtrees. |
GISAID doesn't give the GenBank accession numbers, so we cannot easily say whether our sample matches theirs. However, the tree we can infer from the complete genomes (high-divergent, non-alignable regions excluded) looks very similar, as shown next, and some of the labels match up.
 |
| Fig. 1 Maximum likelihood (ML) tree inferred for our sample using (old, v.8.0.20) RAxML. Roman numbers refer to earlier defined Types 1–9 (Tree and viruses – the SARS group), Arabic numbers give nonparametric bootstrap (BS) support based on 100 BS pseudoreplicates (number of neccessary BS replicates determined by the extended majority rule criterion). Branches without Arabic number are unambiguous (BS = 100). |
Most importantly, all but three branches have unambiguous support: the phylogeny of this sample is resolved. Unfortunately, as our recurring readers already know, this nearly resolved tree simplifies a much more complex situation.
 |
| The Neighbor-net with recombinations and mutational trends (arrows, connectives; cf. Tree and viruses – the SARS group). |
Hack and slash
A simple method to fish for different evolutionary histories in a genome is to cut the virus genomes into sub-sequences, infer a tree for each sub-sequence, and then compare the trees. Most researchers compare trees by showing them and discussing which one makes most sense. Here is an example from Corman et al. (2014), who searched for the root of MERS (Middle East Respiratory Syndrome) virus, an illness closely related to SARS.
 |
| Reprint of Corman et al. 2014, fig. 3 with colors added to EriCoV (green) and HKU/BtCoV (olive) groups |
Each tree in their Fig. 4A and B (Bayesian majority rule consensus trees) was inferred from a different part of the genome. Corman et al.'s focus was to root the MERS viruses by identifying a better outgroup. However, note that the new sister-group (red, green stars – sister to MERS; orange stars – sister to someone else) moves, and so does the green EriCoV clade and the olive HKU/BtCoV group (clade in some trees and grade in others). Do some of these trees get it wrong? Or is, eg. NeoCoV the product of reticulate evolution (here: ancient recombination)? Some parts of its genome might be derived from a common ancestor with MERS (blues), and others from a common ancestor with KW2E (black) and EriCoV (green).
Our complete matrix has 27,333 characters, providing nearly 6,000 distinct alignment patterns (abbreviated DAP, below), which is a lot — the GISAID link above also provides a graphical representation of site divergence. However, probabilistic tree inference methods (ML, Bayes) can handle moderate to high levels of divergence in the data. On the other hand, they also need a certain amount of data to perform well (see also: Inferring a tree with 12000 [or more] virus genomes). So, for my experiment, I hacked the matrix into nine bits of equal size, ie. each submatrix has a bit more than 3,000 nucleotides, providing between 615 (bit #5) and 1029 (bit #1) DAPs.
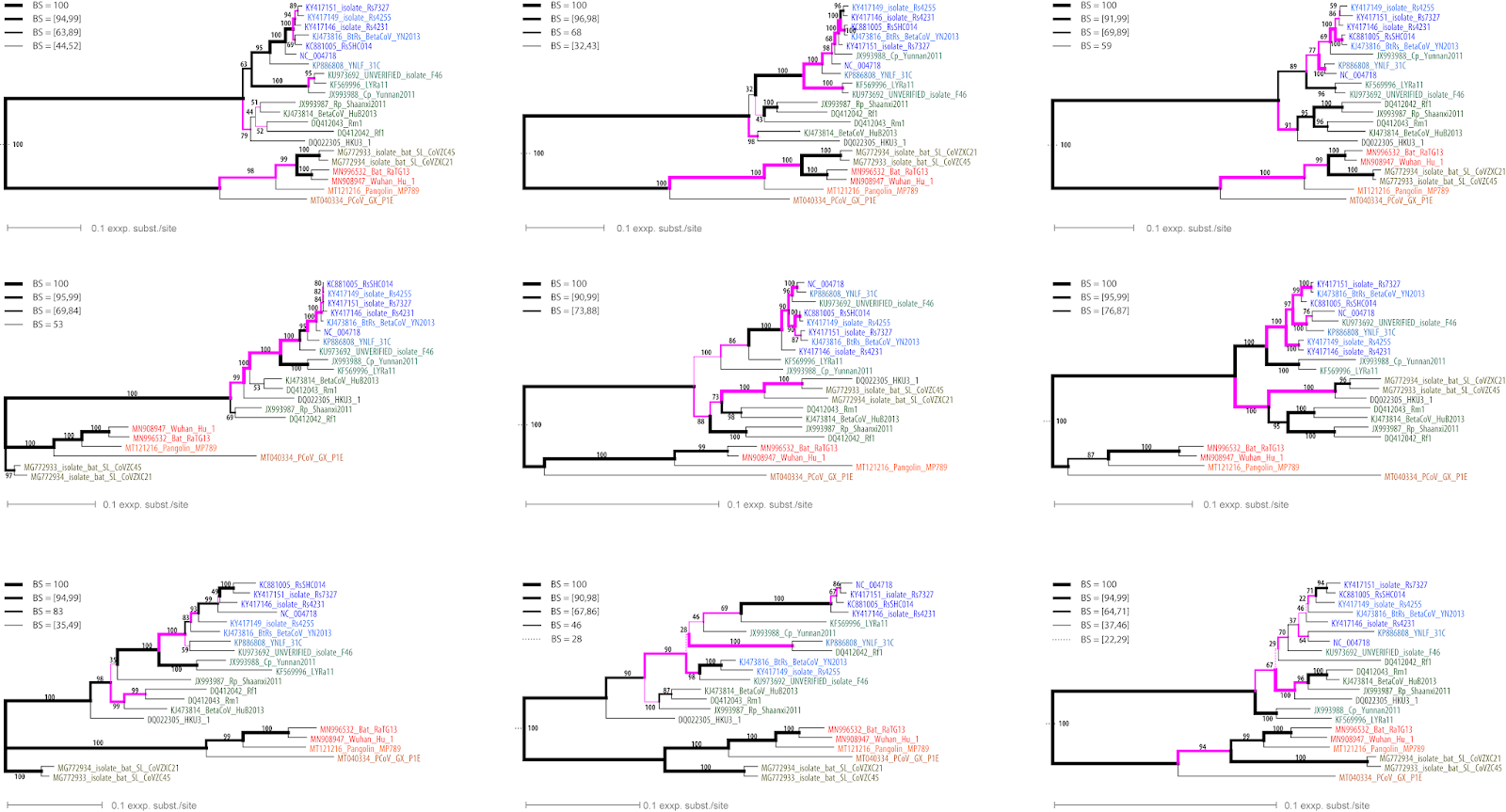 |
| Fig. 2 Nine ML trees with BS support annotated along branches, each based on a ~3000 nucleotide long bit of the genomes (ordered left-right, top-bottom). Purple highlights branches conflicting with the complete genome tree. |
Our nine trees (shown above) are not badly resolved, as most branches get substantial support. But they are not congruent. If we are dealing with recombination, then we might assume that all of these trees do show an actual aspect of the evolutionary history of the genomes. That is, they are all right and wrong, at the same time.
Moreover, we have high supported clades conflicting with the complete genome tree's (Fig. 1) topology. The signal issues, due to recombination (see Trees and viruses...), did not decrease branch support. That is, 6,000+ DAP is a lot, and recombination only affects a part of the complete genome, possibly quite a small part.
Non-trivial evolution needs more than trivial graphs
To depict the reticulate phylogeny of the virus sample, we need to consider the differences seen in the hacked-and-slashed matrix trees. This can easily be illustrated using a network, instead of a set of trees, as shown here.
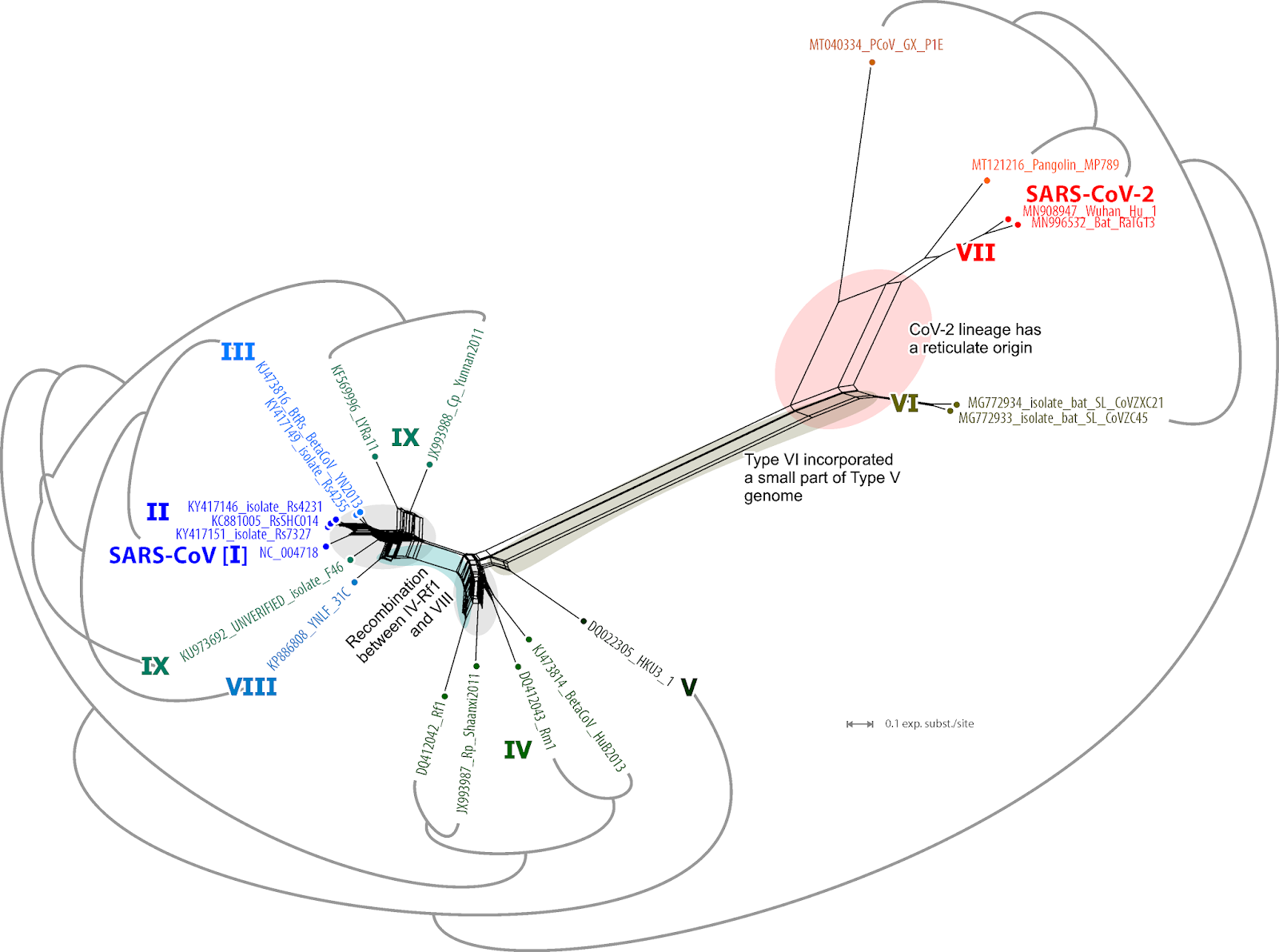 |
| Fig. 3 A (strict) consensus network of all nine trees, in which the edge lengths give the sum of the branch lenghts in the tree sample. The gray brackets give the topology of the near-fully resolved complete genome tree. |
The graph above is a phylogenetic network: the competing edge bundles represent the different inferred histories of bits of the genomes. The SARS-CoV-2 lineage seems to be the product of (ancient) recombination, and recombination also played a role in forming the members of the original SARS-CoV group.
 |
| Fig. 4 Pruned consensus network showing only the CoV(-1) lineage exhibiting various levels of recombination within and between clades as defined by the complete genome tree (tree sample sames as in Fig. 3). |
Consensus networks can also be used to summarize the support for alternative splits, as shown next.
 |
| Fig. 5 Sum-support consensus network based on the bit-wise BS analyses (111/112 pseudoreplicates generated per bit). Only splits are shown occurring in at least 20% of all BS replicates, i.e. splits supported by at least two bits, trivial splits are collapsed. Colored splits represent according groups/clades in the full-genome tree (Fig. 1). Inlet: 'splits rose' showing competing splits patterns within Types II and III (cf. according subtrees/-trunks in Fig. 2 and Fig. 4). |
In contrast to the networks before (Figs. 3, 4), generated using the same algorithm*, the BS consensus network in Fig. 5 is not a phylogenetic network. The boxes don't reflect disparate histories of parts of the genomes but the varying support for competing topological alternatives. By summing up the bit-wise BS analyses instead of bootstrapping the entire data (the BS consensus network for the full data, Fig. 1, shows only two boxes), we get a better idea which aspects of the all-genome tree find robust support across the genome.**
Conclusion
Sub-dividing an alignment is a really quick† way to fish for evidence of recombination, especially when one then uses a consensus network to summarise the resulting partial trees.
For interpretation, a tree is a very simple, trivial, and hence appealing graph: A is sister to B and so on. Even a child can interpret a tree. Networks are already visually more challenging, but whenever an organism's evolution doesn't follow a tree (as for viruses), we shouldn't use a tree to depict its phylogeny (or reconstruct its evolution).
Data availability
The dataset used for our experiment is a taxon subset of the original data set, available via figshare (with a permanent, hence, citable DOI):
Grimm GW, Morrison D. 2020. Harvest and phylogenetic network analysis of SARS virus genomes (CoV-1 and CoV-2). figshare Dataset. https://doi.org/10.6084/m9.figshare.12046581
References
Corman VM, Ithete NL, Richards LR, Schoeman MC, Preiser W, Drosten C, Drexlera JF (2014) Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African Bat. Journal of Virology 88: 11297–11303.
* SplitsTree includes five options to determine "edge weights" (= edge-lengths) in case of Consensus networks: "median" and "mean" average the branch-lengths in the tree sample; "count", the setting used to generate Support consensus networks, counts how often a certain taxon bipartition (split) is found in the tree sample – an edge length is proportional to the frequency of a split; "sum", used here to generate the first network, summarizes the branch-lengths; and "none" discards both branch-lengths and split frequency.
** A split supported only by one of the nine bits, even if unambiguous, ie. present in all 111 (112) per bit BS replicates, will not be represented in the sum-Support consenus network using a cut-off of 20%.
† The complete set of ML analyses took 20 min on a stand-alone computer; consensus networks are generated in a blink, and take hardly a minute even when using trees with many leaves.