At this year's
Society for Molecular Biology and Evolution meeting in Lyon I presented ongoing work estimating the distribution of fitness effects, which is a collaborative venture with
Molly Przeworski and
Peter Andolfatto. Earlier versions of this research appeared in talks I presented at Chicago in December (Ecology and Evolution Departmental seminar) and Liverpool in January (UK Population Genetics Group meeting), and it follows on from last year's SMBE presentation in which I discussed methods to tease out sub-genic variation in selection pressure.
There is intrinsic interest in the fitness effects of novel mutations in coding regions of the genome, especially the relative frequency of occurrence of neutral, beneficial and deleterious variants. Yet estimating the
distribution of fitness effects (the DFE) is also of practical use when localizing the signal of adaptive evolution. The reason is that in Bayesian analyses, the assumed DFE can influence the strength of evidence for or against adaptation at a particular site. Consequently it is preferably to estimate the DFE at the same time as detecting adaptation at individual sites to avoid prior assumptions unduly influencing the results.
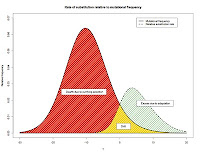
Having estimated the DFE, it is of use in quantifying the relative contribution of adaptation versus drift to genome evolution. The figure, taken from my talk in Lyon (
slides here), illustrates the idea when a normal distribution is used to estimate the DFE; the relative area of the green to the yellow shaded regions represents the respective contribution of adaptation versus drift in amino acid substitutions accrued along the
Drosophila melanogaster lineage.



