Spermatophyte morphological matrices that combine extinct and extant taxa notoriously have low branch support, as traditionally established using non-parametric bootstrapping under parsimony as optimality criterion. Coiro, Chomicki & Doyle (2017) recently published a
pre-print to show that this can be overcome to some degree by changing to Bayesian-inferred posterior probabilities. They also highlight the use of support consensus networks for investigating potential conflict in the data. This is a good start for a scientific community that so far has put more of their trust in either (i) direct visual comparison of fossils with extant taxa or (ii) collections of most parsimonious trees inferred based on matrices with high level of probably homoplasious characters and low compatibility. But do those matrices really require or support a tree? Here, I try to answer this question.
BackgroundCoiro et al. mainly rely on a recent matrix by Rothwell & Stockey (2016), which marks the current endpoint of a long history of putting up and re-scoring morphology-based matrices (Coiro et al.’s fig. 1b). All of these matrices provide, to various degrees, ambiguous signal. This is not overly surprising, as these matrices include a relatively high number of fossil taxa with many data gaps (due to preservation and scoring problems), and combine taxa that perished a hundred or more millions years ago with highly derived, possibly distant-related modern counterparts.
Rothwell & Stockey state (p. 929) "
As is characteristic for the results from the analysis of matrices with low character state/taxon ratios, results of the bootstrap analysis (1000 replicates) yielded a much less fully resolved tree (not figured)." Coiro et al.’s consensus trees and network based on 10,000 parsimony bootstrap replicates nicely depicts this issue, and may explain why Rothwell & Stockey decided against showing those results. When studying an earlier version of their matrix (Rothwell, Crepet & Stockey 2009), they did not provide any support values, citing a paper published in 2006, where the authors state (Rothwell & Nixon 2006, p. 739): “
… support values, whether low or high for particular groups, would only mislead the reader into believing we are presenting a proposed phylogeny for the groups in question. Differences among most-parsimonious trees are sufficient to illuminate the points we wish to make here, and support values only provide what we consider to be a false sense of accuracy in these assessments”.
Do the data support a tree?The problem is not just low support. In fact, the tree showed by Rothwell & Stockey with its “pectinate arrangement” conflicts in parts with the best-supported topology, a problem that also applied to its 2009 predecessor. This general “pectinate” arrangement of a large, low or unsupported grade is not uncommon for strict consensus trees based on morphological matrices that include fossils and extant taxa (see e.g. the more proximal parts of the Tree of Life, e.g. birds and their dinosaur ancestors).
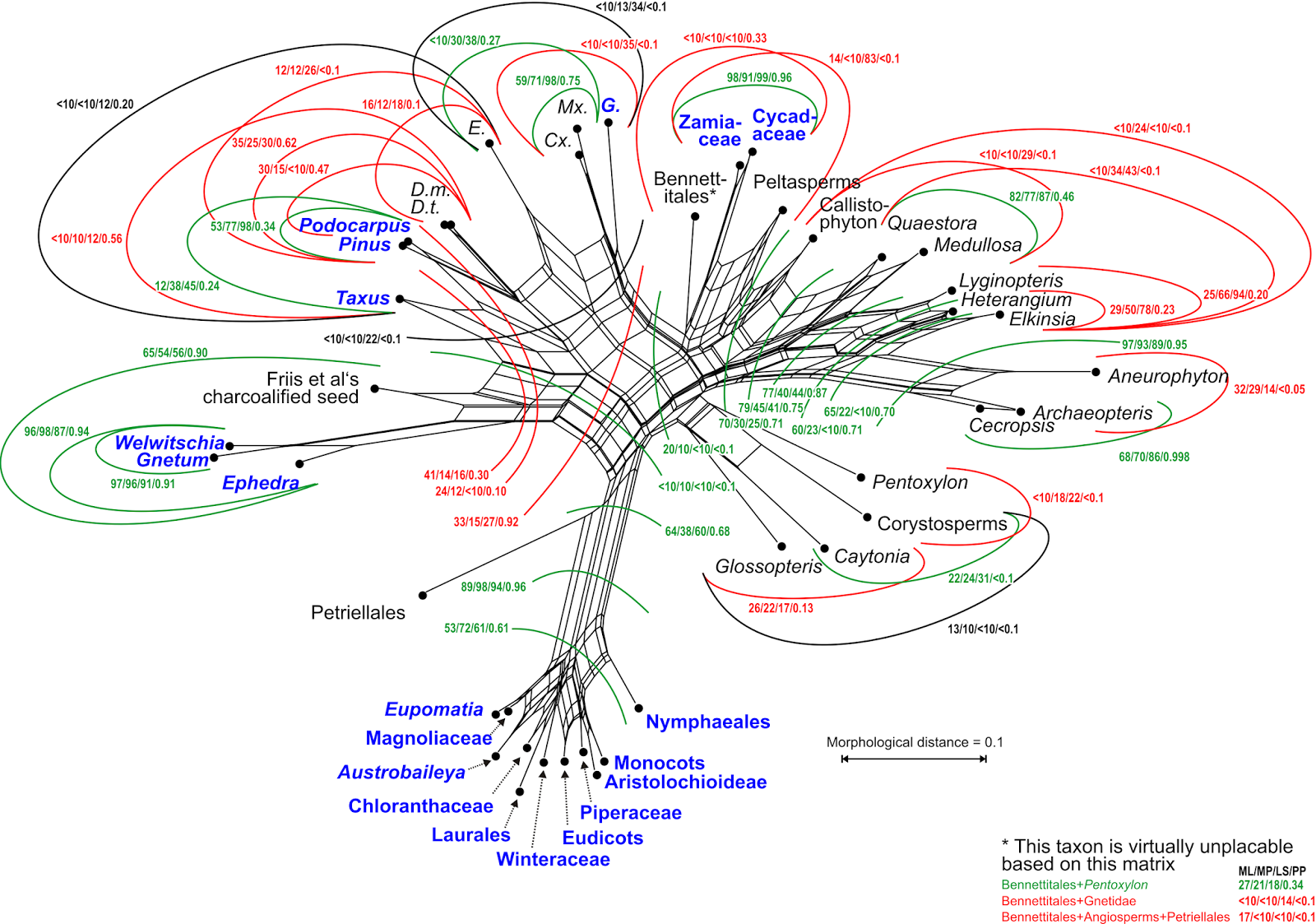
The support patterns indicate that some of the characters are compatible with the tree, but many others are not. Of the 34 internodes (branches) in the shown tree (their fig. 28 shows a strict consensus tree based on a collection of equally parsimonious trees), 12 have lower bootstrap support under parsimony than their competing alternatives (
Fig. 1). Support may be generally low for any alternative, but the ones in the tree can be among the worst.
The main problem is that the matrix simply does not provide enough tree-like signal to infer a tree. Delta Values (Holland et al. 2002) can be used as a quick estimate for the treelikeliness of signal in a matrix. In the case of large all-spermatophyte matrices (Hilton & Bateman 2006; Friis et al. 2007; Rothwell, Crepet & Stockey 2009; Crepet & Stevenson 2010), the matrix Delta Values (mDV) are ≥ 0.3. For comparison, molecular matrices resulting in more or less resolved trees have mDV of ≤ 0.15. The individual Delta Values (iDV), which can be an indicator of how well a taxon behaves during tree inference, go down to 0.25 for extant angiosperms – very distinct from all other taxa in the all-spermatophyte matrices with low proportions of missing data/gaps – and reach values of 0.35 for fossil taxa with long-debated affinities.
The newest 2016 matrix is no exception with a mDV of 0.322 (the highest of all mentioned matrices), and iDVs range between 0.26 (monocots and other extant angiosperms) and 0.39 for
Doylea mongolica (a fossil with very few scored characters). In the original tree,
Doylea (represented by two taxa) is part of the large grade and indicated as the sister to Gnetidae (or Gnetales) + angiosperms (molecular trees associate the Gnetidae with conifers and
Ginkgo). According to the bootstrap analysis,
Doylea is closest to the extant Pinales, the modern conifers. Coiro et al. found the same using Bayesian inference. Their posterior probability (PP) of a
Doylea-Podocarpus-Pinus clade is 0.54, and Rothwell & Stockey’s
Doylea-Ginkgo-angiosperm clade conflicts with a series of splits with PPs up to 0.95.
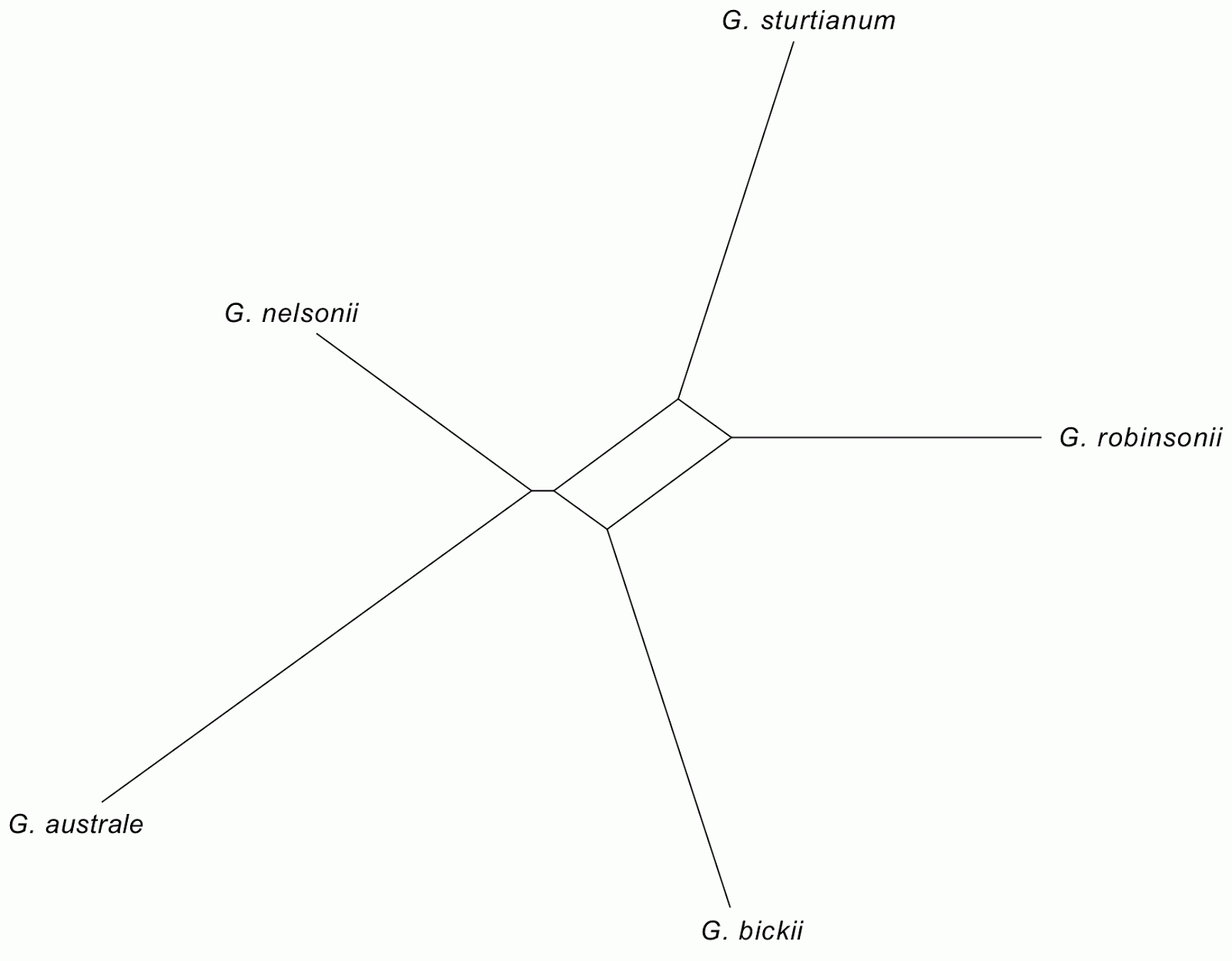
 |
Figure 1. Parsimony bootstrap network based on 10,000 pseudoreplicate trees
inferred from the matrix of Rothwell & Stockey.
Edges not found in the authors’ tree in red, edges also found in the tree in green.
Extant taxa in blue bold font. The edge length is proportional to the frequency of the
according split (taxon bipartition, branch in a possible tree) in the pseudoreplicate
tree sample. The network includes all edges of the authors’ tree except for
Doylea + Gnetidae + Petriellales + angiosperms vs. all other gymnosperms and
extinct seed plant groups. Such a split has also no bootstrap support (BS < 10)
using least-square and maximum likelihood optimum criteria. |
Do the data require a tree?As David made a point in an earlier
post, neighbour-nets are not really “phylogenetic networks” in the evolutionary sense. Being unrooted and 2-dimensional, they don’t depict a phylogeny, which has to be a sort of (rooted) tree, a one-dimensional graph with time as the only axis (this includes reticulation networks where nodes can be the crossing point of two internodes rather than their divergence point). The neighbour-net algorithm is an extension into two dimensions of the neighbour-joining algorithm, the latter infers a phylogenetic tree serving a distance criterion such as minimum evolution or least-squares (Felsenstein 2004). Essentially, the neighbour-net is a ‘meta-phylogenetic’ graph inferring and depicting the best and second-best alternative for each relationship. Thus, neighbour-nets can help to establish whether the signal from a matrix, treelike or not as it is the cases here, supports potential and phylogenetic relationships, and explore the alternatives much more comprehensively than would be possible with a strict-consensus or other tree (
Fig. 2).
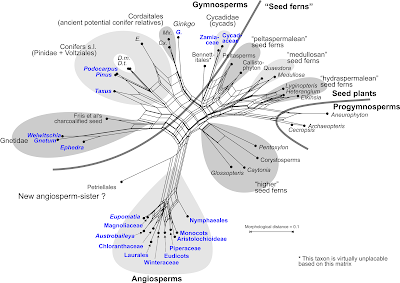 |
Figure 2. Neighbour-net based on a mean distance matrix inferred
from the matrix of Rothwell & Stockey.
The distance to the "progymnosperms", a potential ancestral group of the
seed plants, can be taken as a measurement for the derivedness of each
major group. The primitive seed ferns are placed between progymnosperms
and the gymnosperms connected by partly compatible edge bundles; the
putatively derived "higher seed ferns" isolated between the progymnosperms
and the long-edged angiosperms. Shared edge-bundles and 'neighbourness'
reflect quite well potential phylogenetic relationships and eventual ambiguities,
as in the case of Gnetidae. Colouring as in Figure 1; some taxon names
are abbreviated. |
In addition, neighbour-nets usually are better backgrounds to map patterns of conflicting or partly conflicting support seen in a bootstrap, jackknife or Bayesian-inferred tree sample. In
Fig. 3, I have mapped the bootstrap support for alternative taxon bipartitions (branches in a tree) on the background of the neighbour-net in
Fig. 2.
Obvious and less-obvious relationships are simultaneously revealed, and their competing support patterns depicted. Based on the graph, we can see (edge lengths of the neighbour-net) that there is a relatively weak primary but substantial bootstrap support for the Petriellales (a recently described taxon new to the matrix) as sister to the angiosperms. Several taxa, or groups of closely related taxa, are characterised by long terminal edges/edge bundles, rooting in the boxy central part of the graph. Any alternative relationship of these taxa/taxon groups receives equally low support, but there are notable differences in the actual values.
There is little signal to place most of the fossil “seed ferns” (extinct seed plants) in relation to the modern groups, and a very ambiguous signal regarding the relationship of the Gnetidae (or Gnetales) with the two main groups of extant seed plants, the conifers (Pinidae; see C. Earle’s
gymnosperm database) and angiosperms (for a list and trees, see P. Stevens’
Angiosperm Phylogeny Website).
The Gnetidae is a strongly distinct (also genetically) group of three surviving genera, being a persistent source of headaches for plant phylogeneticists. Placed as sister to the Pinaceae (‘Gnepine’ hypothesis) in early molecular trees (long-branch attraction artefact), the currently favoured hypothesis (‘Gnetifer’) places the Gnetidae as sister to all conifers (Pinatidae) in an all-gymnosperm clade (including
Gingko and possibly the cycads).
As favoured by the branch support analyses, and contrasting with the preferred 2016 tree, the two
Doyleas are placed closest to the conifers, nested within a commonly found group including the modern and ancient conifers and their long-extinct relatives (Cordaitales), and possibly
Ginkgo (Ginkgoidae). In the original parsimony strict consensus tree, they are placed in the distal part as sister to a Gnetidae and Petriellales + angiosperms (possibly long-branch attraction). The grade including the ‘primitive seed ferns’ (
Elkinsia through
Callistophyton), seen also in Rothwell and Stockey’s 2016 tree, may be poorly supported under maximum parsimony (the criterion used to generate the tree), but receives quite high support when using a probabilistic approach such as maximum likelihood bootstrapping or Bayesian inference to some degree (
Fig. 3; Coiro, Chomicki & Doyle 2017).
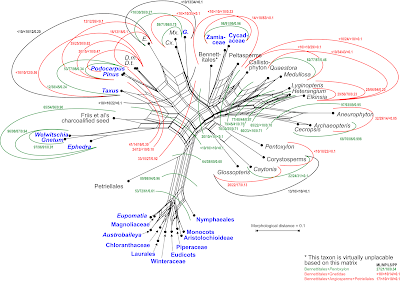 |
Figure 3. Neighbour-net from above used to map alternative support patterns.
Numbers refer to non-parametric bootstrap (BS) support for alternative phylogenetic
splits under three optimality criteria: maximum likelihood (ML) as implemented in
RAxML (using MK+G model), maximum parsimony (MP), and least-squares
(via neighbour-joining, NJ; using PAUP*); and Bayesian posterior probabilties
(using MrBayes 3.2; see Denk & Grimm 2009, for analysis set-up). The circular
arrangement of the taxa allows tracking most edges in the authors’ tree and their,
sometimes better supported, alternatives. The edge lengths provide direct
information about the distinctness of the included taxa to each other; the structure
of the graph informs about the how tree-like the signal is regarding possible
phylogenetic relationships or their alternatives. Colouring as in Figure 1;
some taxon names are abbreviated. |
Numerous morphological matrices provide non-treelike signals. A tree can be inferred, but its topology may be only one of many possible trees. In the framework of total evidence, this may be not such a big problem, because the molecular partitions will predefine a tree, and fossils will simply be placed in that tree based on their character suites. Without such data, any tree may be biased and a poor reflection of the differentiation patterns.
By not forcing the data in a series of dichotomies, neighbour-nets provide a quick, simple alternative. Unambiguous, well-supported branches in a tree will usually result in tree-like portions of the neighbour net. Boxy portions in the neighbour-net pinpoint the ambiguous or even problematic signals from the matrix. Based on the graph, one can extract the alternatives worth testing or exploring. Support for the alternatives can be established using traditional branch support measures. Since any morphological matrix will combine those characters that are in line with the phylogeny as well as those that are at odds with it (convergences, character misinterpretations), the focus cannot be to infer a tree, but to establish the alternative scenarios and the support for them in the data matrix.
ReferencesCoiro M, Chomicki G, Doyle JA. 2017. Experimental signal dissection and method sensitivity analyses reaffirm the potential of fossils and morphology in the resolution of seed plant phylogeny.
bioRxiv DOI:10.1101/134262
Crepet WL, Stevenson DM. 2010. The Bennettitales (Cycadeoidales): a preliminary perspective of this arguably enigmatic group. In: Gee CT, ed.
Plants in Mesozoic Time: Morphological Innovations, Phylogeny, Ecosystems. Bloomington: Indiana University Press, pp. 215-244.
Denk T, Grimm GW. 2009. The biogeographic history of beech trees.
Review of Palaeobotany and Palynology 158: 83-100.
Felsenstein J. 2004.
Inferring Phylogenies. Sunderland, MA, U.S.A.: Sinauer Associates Inc.
Friis EM, Crane PR, Pedersen KR, Bengtson S, Donoghue PCJ, Grimm GW, Stampanoni M. 2007. Phase-contrast X-ray microtomography links Cretaceous seeds with Gnetales and Bennettitales.
Nature 450: 549-552 [all important information needed for this post is in the supplement to the paper; a figure showing the actual full analysis results can be found at
figshare]
Hilton J, Bateman RM. 2006. Pteridosperms are the backbone of seed-plant phylogeny.
Journal of the Torrey Botanical Society 133: 119-168.
Holland BR, Huber KT, Dress A, Moulton V. 2002. Delta Plots: A tool for analyzing phylogenetic distance data.
Molecular Biology and Evolution 19: 2051-2059.
Rothwell GW, Crepet WL, Stockey RA. 2009. Is the anthophyte hypothesis alive and well? New evidence from the reproductive structures of Bennettitales.
American Journal of Botany 96: 296–322.
Rothwell GW, Nixon K. 2006. How does the inclusion of fossil data change our conclusions about the phylogenetic history of the euphyllophytes?
International Journal of Plant Sciences 167: 737–749.
Rothwell GW, Stockey RA. 2016. Phylogenetic diversification of Early Cretaceous seed plants: The compound seed cone of
Doylea tetrahedrasperma.
American Journal of Botany 103: 923–937.
Schliep K, Potts AJ, Morrison DA, Grimm GW. 2017. Intertwining phylogenetic trees and networks.
Methods in Ecology and Evolution DOI:10.1111/2041-210X.12760.