It has been noted before that we have a wide range of mathematical techniques available for producing data-display networks, most notably the many variants of splits graphs (see Huson & Scornavacca 2011). For example, NeighborNets and Consensus networks are commonly encountered in the phylogenetics literature, and Reduced median networks and Median-joining networks are commonly used for haplotype networks in population biology.
However, there are few techniques used to produce evolutionary networks. Studies of reticulate evolutionary histories, which include recombination networks, hybridization networks, introgression networks and HGT networks, have no unifying theme as yet. So, the biological literature has many papers in which biologists struggle with reticulate evolutionary histories using ad hoc collections of techniques, which often boil down to simply presenting incongruent phylogenetic trees from different datasets (see Morrison 2014a).
So, maybe a brief look at the current state of play with evolutionary networks would be useful. There are enough worthwhile techniques out there for people to be using them more often than they are.
AssumptionsAlmost all current phylogenetic methods assume that the basic building unit is a non-recombining sequence block, for which the evolutionary history is strictly tree-like. We tend to call these blocks "genes" and their history "gene trees", but this is just for semantic convenience. In practice, we first collect data for various loci, and we then simply make the assumption that there is recombination between the loci but not within them. This is basically the assumption of independence between loci. At the limit, each nucleotide along a chromosome has a tree-like history, but for aggregations of nucleotides it is all assumptions.
Furthermore, we assume that there are no data errors that will confound any reconstruction of the phylogenetic trees. Possible sources of error include: incorrect data (e.g. contamination), inappropriate sampling (taxa or characters), and model mis-specification. Any of these errors will lead to stochastic variation at best and to bias at worst.
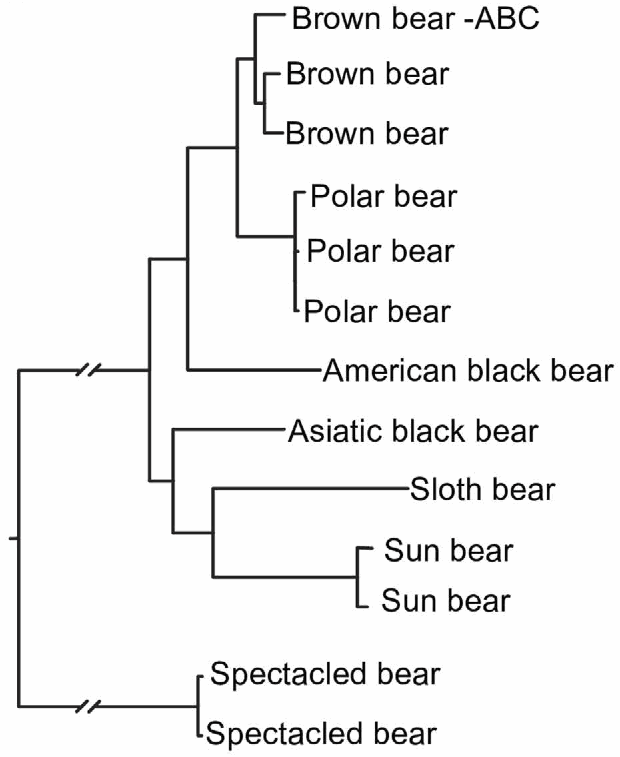
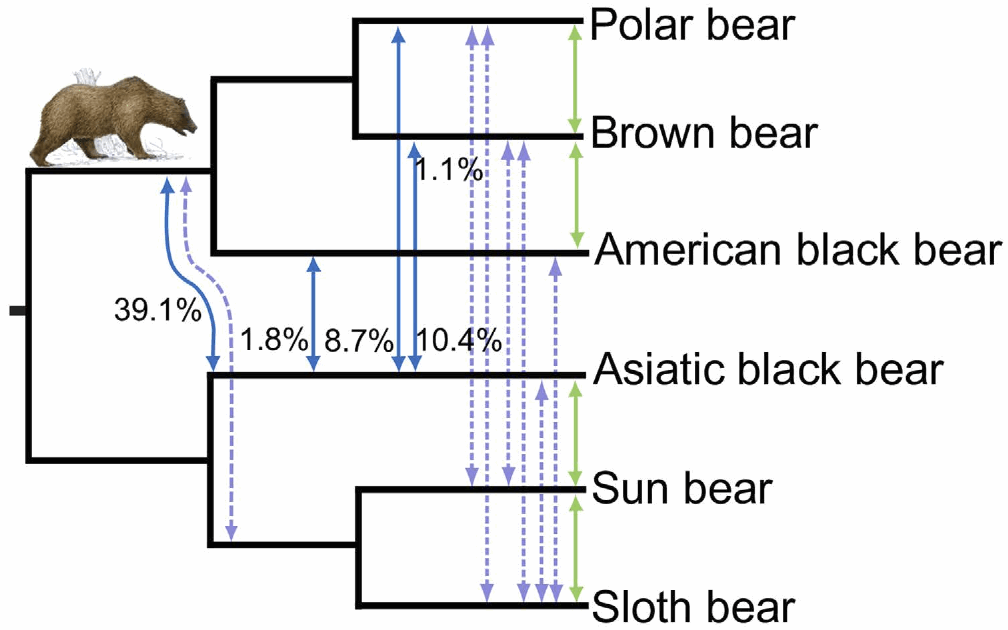
Gene-tree incongruenceReticulate evolutionary processes lead to gene trees that are not all congruent. However, there are two other processes that have been widely recognized as also producing gene-tree incongruence, but which do not involve reticulation in the strict sense: incomplete lineage sorting (deep coalescence; ancestral polymorphism), and gene duplication-loss.
Many studies have now shown that stochastic variation due to ILS can be very large (see Degnan & Rosenberg 2009), and that this varies in relation to both the population sizes of the taxa and the times between divergence events. The expectation of completely congruent gene trees is thus very naive, even when the evolutionary history of the taxa has been strictly tree-like. A number of methods have been developed to reconstruct species trees in the face of ILS (Nakhleh 2013).
DL involves gene duplication (which can be repeated to create gene families) followed by selective gene loss. The phylogenetic history of the genes is usually presented as an unfolded species tree, where each gene copy has its own part of the tree. A number of methods have been developed to reconstruct gene DL histories given a "known" species tree, which is called gene-tree reconciliation (Szöllősi et al 2015). However, our interest here is in the reverse process, in which reconstructed but incongruent gene trees are combined into a single species tree, given a model of duplication and selective loss, which is called species-tree inference (which is the same as cophylogeny reconstruction; Drinkwater & Charleston 2014).
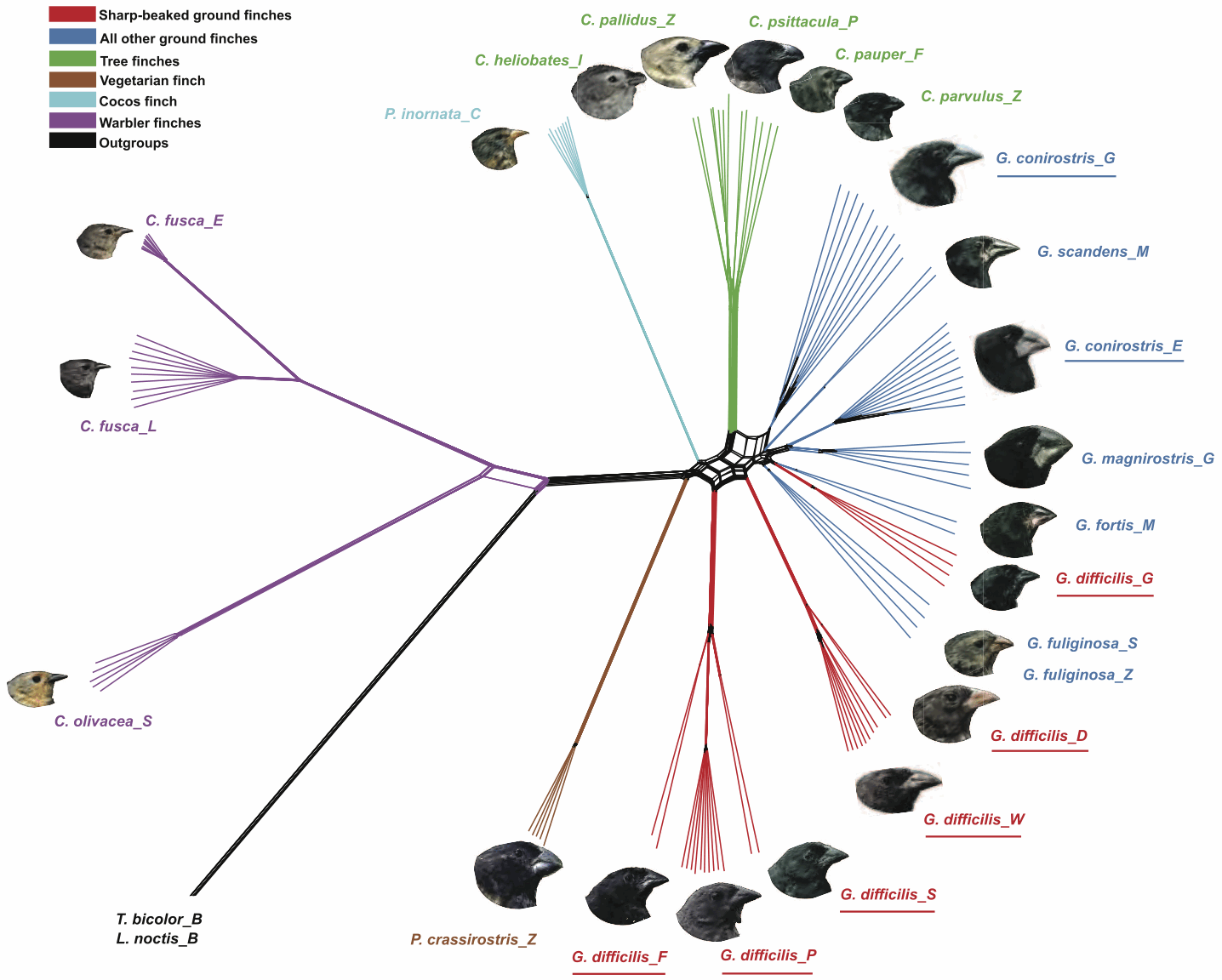
ReticulationsKnown biological processes such as recombination, reassortment, hybridization, introgression and horizontal gene transfer all create reticulate phylogenetic histories. However, it is a moot point as to whether these processes can be distinguished from each other solely in the context of an evolutionary network (Holder et al. 2001; Morrison 2015). These evolutionary processes operate by distinct biological mechanisms, but the evolutionary patterns that they create can all be rather similar. The processes all result in gene flow among contemporaneous organisms (usually called horizontal flow or transfer), whereas other evolutionary processes involve gene flow from parent to offspring (usually called vertical inheritance), including ILS and DL. These gene flows create incongruent gene histories, which we may detect directly in the data or via reconstructed gene trees. The patterns of incongruence do not necessarily allow us to infer the causal process.
There are a number of differences in pattern, but the consistency of these is doubtful. Polyploid hybridization produces the most distinctive pattern, because there is duplication of the genome in the hybrid. However, subsequent aneuploidy will serve to obscure this pattern. Homoploid hybridization nominally involves 50% of the genome coming from difference sources, while introgression ultimately involves a smaller percentage. However, in practice, genome mixtures vary continuously from 0 to 50%. HGT also involves a small percentage of the genome, but in theory it also can vary from 0 to 50%. Reassortment produces mixtures of viral genes, which can occur in such a great number that reconstructing the history is severely problematic.
So, in the absence of independent experimental evidence, distinguishing one form of evolutionary network from another is almost a matter of definition. This has become increasingly obvious in the methodological literature, where semantic confusion abounds.
For example, a network produced directly from a set of characters has usually been called a "recombination network", while one produced from a set of trees has usually been called a "hybridization network", irrespective of what processes the gene trees represent. Furthermore, models that add reticulation events to DL trees have usually referred to the horizontal gene flow as "HGT", whereas models that add reticulation events to ILS trees have usually referred to the horizontal gene flow as "hybridization" (Morrison 2014a). Studies of horizontal gene flow during human evolution have usually referred to "admixture", which is a more process-neutral term.
In many, if not most, cases we might all be better off if network methods simply distinguish gene flow among contemporaries (horizontal) from gene inheritance between generations (vertical), rather than trying to infer a process — process inference can often best take place after network construction. This does not help anthropologists, of course, who are dealing with evolutionary networks where oblique gene flow is possible (so that they do not have
Time inconsistency in evolutionary networks).
MethodsThere seems to be a dichotomy of purposes to current method development, which are neatly summarized by the contrasting theoretical views of Mindell (2013) and Morrison (2014b). These views each recognize that evolutionary history involves both vertical and horizontal processes, but they reconstruct the resulting evolutionary patterns as a species tree and a species network, respectively. Obviously, this blog is dedicated to the latter point of view, but it is the former one (the so-called Tree of Life) that seems to currently dominate the literature.
Focussing on gene-tree inference, Szöllősi et al (2015) provide a comprehensive review of the various models that have been used to describe the dependence between gene trees and species trees. Essentially, gene trees are contained within the species tree, and they may differ from it in relative branch lengths and/or topology. The differences between genes and species are the result of population-level processes, often modeled using the coalescent. These authors recognize four current classes of probabilistic model that combine different evolutionary processes:
- the DLCoal model, which combines coalescence and DL
- the DTLSR model and the ODT model, both of which combine gene transfer and DL
- models that combine hybridization and ILS
- models of allopolyploidization.
When inferring species trees from gene trees (species-tree inference), we basically combine the scores for all of the gene trees, and then search for the species tree with the best overall score. This involves adding the scores in parsimony analyses, or multiplying the conditional probabilities in likelihood analyses (ie. maximum-likelihood or bayesian context). Many methods have been developed for inferring a species tree based on multi-locus data. These differ in whether the gene and species trees are estimated simultaneously or sequentially, and in how the gene trees are used to infer the species tree. Nakhleh (2013) and Szöllősi et al (2015) discuss both parsimony and likelihood methods for species-tree inference based on either ILS or DL models.
Extending these ideas to infer networks (rather than species trees) is a bit more tricky, and most of the work to date has involved combining hybridization and ILS. There has been no recent summary of the ideas. However, calculating the parsimony score of a network, given a set of gene-tree topologies, has been beed addressed by Yu et al (2011); and Yu et al (2013a) have extended these ideas to heuristically search the network space for the optimal network (the one that minimizes the number of extra reticulation lineages in a species tree). Furthermore, methods for computing the likelihood of a phylogenetic network, given a set of gene-tree topologies, have been devised by Yu et al (2012, 2013b); and Yu et al (2014) have extended these ideas to heuristically search for the maximum-likelihood network for limited cases of introgression or hybridization (since they differ only in degree).
There are also several methods that simply use gene-tree incongruence to infer reticulation events in a species network (Huson et al. 2010). Basically, these methods combine gene trees into "hybridization networks" by minimizing the number of reticulations required for reconciliation, measured either by counting the reticulations or calculating the network level. The combinatorial optimization can be based on trees, triplets or clusters, using parsimony as the optimality criterion. These methods model homoploid hybridization by assuming that reticulation is the sole cause of all gene-tree incongruence. This means that they are likely to overestimate the amount of reticulation in a dataset when other processes are co-occurring.
The most completely developed network methods involve data for allopolyploid hybrids. Here, there are multiple copies of each gene, one in each copy of the genome, so that allopolyploid hybrids have more copies than do their diploid parent taxa. To construct a hybridization network topology, Huber et al (2006) developed a parsimony method based on first estimating a multi-labeled gene tree, and then searching for the single-labeled network that best accommodates the multiple gene patterns. The model has been extended to heuristically include ILS (Marcussen et al 2012), as well as dates for the internal nodes (Marcussen et al 2015). Jones et al. (2013) have also developed models that incorporate ILS in a bayesian context, but only for the case of a single hybridization event between two diploid species (an allotetraploid).
Species-tree inference for a pair of gene phylogenies that may be networks not trees, has been considered in terms of parsimony by Drinkwater & Charleston (2014).
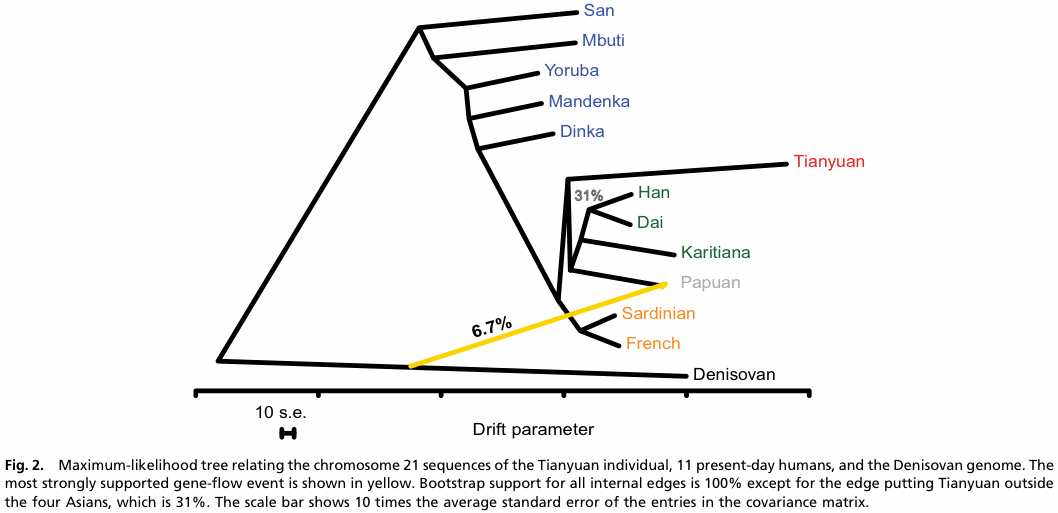
This brings us to the matter of introgression. The massive recent influx of genome-scale data for hominids has lead to the development of methods explicitly for the analysis of what is termed admixture among the lineages. These methods basically work by constructing a phylogenetic tree that includes admixture events, the topology inference being based on allele frequencies. There has been no formal comparison of the methods, and not much application to non-humans. Three such methods have been produced so far (Patterson et al 2012; Pickrell & Pritchard 2012; Lipson et al 2013).
Recombination has somewhat been the poor cousin to other causes of reticulation, as most network methods assume it to be absent. Nevertheless, Gusfield (2014) has recently provided an ample survey of the study methods available to date.
ReferencesDegnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent.
Trends in Ecology & Evolution 24: 332-340.
Drinkwater B, Charleston MA (2014) An improved node mapping algorithm for the cophylogeny reconstruction problem.
Coevolution 2: 1-17.
Gusfield D (2014)
ReCombinatorics: the Algorithmics of Ancestral Recombination Graphs and Explicit Phylogenetic Networks. MIT Press, Cambridge.
Holder MT, Anderson JA, Holloway AK (2001) Difficulties in detecting hybridization.
Systematic Biology 50: 978-982.
Huber KT, Oxelman B, Lott M, Moulton V (2006) Reconstructing the evolutionary history of polyploids from multilabeled trees.
Molecular Biology & Evolution 23: 1784-1791.
Huson D, Rupp R, Scornavacca C (2010)
Phylogenetic Networks: Concepts, Algorithms, and Applications. Cambridge University Press, Cambridge.
Huson DH, Scornavacca C (2011) A survey of combinatorial methods for phylogenetic networks.
Genome Biology & Evolution 3: 23-35.
Jones G, Sagitov S, Oxelman B (2013) Statistical inference of allopolyploid species networks in the presence of incomplete lineage sorting.
Systematic Biology 62: 467-478.
Lipson M, Loh P-R, Levin A, Reich D, Patterson N, and Berger B (2013) Efficient moment-based inference of population admixture parameters and sources of gene flow.
Molecular Biology & Evolution 30: 1788-1802.
Marcussen T, Heier L, Brysting AK, Oxelman B, Jakobsen KS (2015) From gene trees to a dated allopolyploid network: insights from the angiosperm genus
Viola (Violaceae).
Systematic Biology 64: 84-101.
Marcussen T, Jakobsen KS, Danihelka J, Ballard HE, Blaxland K, Brysting AK, Oxelman B (2012) Inferring species networks from gene trees in high-polyploid north American and Hawaiian violets (
Viola, Violaceae). S
ystematic Biology 61: 107-126.
Mindell DP (2013) The Tree of Life: metaphor, model, and heuristic device.
Systematic Biology 62: 479-489.
Morrison DA (2014a) Phylogenetic networks: a review of methods to display evolutionary history.
Annual Research and Review in Biology 4: 1518-1543.
Morrison DA (2014b) Is the Tree of Life the best metaphor, model or heuristic for phylogenetics?
Systematic Biology 63: 628-638.
Morrison DA (2015, in press) Pattern recognition in phylogenetics: trees and networks. In: Elloumi M, Iliopoulos CS, Wang JTL, Zomaya AY (eds)
Pattern Recognition in Computational Molecular Biology: Techniques and Approaches. Wiley, New York.
Nakhleh L (2013) Computational approaches to species phylogeny inference and gene tree reconciliation.
Trends in Ecology & Evolution 28: 719-728.
Patterson NJ, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D (2012) Ancient admixture in human history.
Genetics 192: 1065-1093.
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data.
PLoS Genetics 8: e1002967.
Szöllősi GJ, Tannier E, Daubin V, Boussau B (2015) The inference of gene trees with species trees.
Systematic Biology 64: e42-e62.
Yu Y, Barnett RM, Nakhleh L (2013a) Parsimonious inference of hybridization in the presence of incomplete lineage sorting.
Systematic Biology 62: 738-751.
Yu Y, Degnan JH, Nakhleh L (2012) The probability of a gene tree topology within
a phylogenetic network with applications to hybridization detection.
PLoS Genetics 8:
e1002660.
Yu Y, Dong J, Liu KJ, Nakhleh L (2014) Maximum likelihood inference of reticulate evolutionary histories.
Proceedings of the National Academy of Sciences of the USA 111: 16448-16453.
Yu Y, Ristic N, Nakhleh L (2013b) Fast algorithms and heuristics for phylogenomics
under ILS and hybridization.
BMC Bioinformatics 14: S6.
Yu Y, Than C, Degnan JH, Nakhleh L (2011) Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting.
Systematic Biology 60: 138-149.


")
")