Category Archives: DAG
Why are splits graphs still called phylogenetic networks?
This is an issue that has long concerned me, and which I think causes a lot of confusion among biologists. A phylogenetic tree is usually a clear concept — to a biologist, it is a diagram that displays a hypothesis of evolutionary history. The expectation, then, is that a phylogenetic network does the same thing for reticulate evolutionary histories. However, this is not true of splits graphs; and so there is potential confusion.
Mathematically, of course, a phylogenetic tree is a directed acyclic line graph. It is usually constructed, in practice, by first producing an undirected graph based on some pattern-analysis procedure, and then nominating one of the nodes or edges as the root (say, by specifying an outgroup). So, the mathematics is not really connected to the biological interpretation. To a mathematician, the tree is a set of nodes connected by directed edges, and the nodes could represent anything at all, as could the edges. It is the biologist who artificially imposes the idea that the nodes represent real historical organisms connected by the flow of evolution — ancestors connected to descendants by evolutionary events.
A phylogenetic network should logically be a generalization of this idea of a phylogenetic tree, adding the possibility of evolutionary relationships due to gene flow, in addition to the ancestor-descendant relationships. This can be done, but it is only partly done by splits graphs.
That is, a splits graph generalizes the idea of an undirected line graph (an unrooted tree), but not a directed acyclic graph (a rooted tree). It follows the same logic of using a pattern-analysis procedure to produce an undirected graph, although the graph can have reticulations, and thus is a network rather than necessarily being a bifurcating tree. However, it is not straightforward to specify a root in a way that will turn this into an acyclic graph. So, in general it does not represent a phylogeny.
Indeed, splits graphs are simply one form of multivariate pattern analysis, along with clustering and ordination techniques, which are familiar as data-display methods in phenetics (see Morrison D.A. 2014. Phylogenetic networks — a new form of multivariate data summary for data mining and exploratory data analysis. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery 4: 296-312). In this sense, it makes no difference whatsoever what the data represent — they can be data used for phylogenetics, or they could be any other form of multivariate data. Indeed, this point is illustrated in many of the posts in this blog, which can be accessed in the Analyses page.
So, unlike unrooted trees, unrooted splits graphs are not a route to producing a phylogenetic diagram. Mind you, they are a very useful form of multivariate data analysis in their own right, and I value them highly as a form of exploratory data analysis. But that doesn't make them phylogenetic networks in the biological sense.
So, isn't it about time we stopped calling splits graphs "phylogenetic networks"? They aren't, to a biologist, so why call them that?
Posted by in DAG, Evolutionary network, Phylogenetic tree, phylogeny, Splits graph
Tree Alignment Graphs and data-display networks
Data-display networks are a means of visualizing complex patterns in multivariate data. One particular use is for displaying the patterns in a set of trees. For example, Consensus Networks and SuperNetworks are splits graphs that display the patterns common to some specified subset of a collection of trees (eg. a set of equally optimal trees, or a set of trees sampled by a bayesian or bootstrap analysis). Alternatively, Parsimony Networks try to simultaneously display all of the trees in a collection of most-parsimonious trees for a single dataset.
Another display method for multiple trees is what has been called a Cloudogram (see the post Cloudograms and data-display networks). These superimpose the set of all trees arising from an analysis, so that dark areas in such a diagram will be those parts where many of the trees agree on the topology, while lighter areas will indicate disagreement.
Yet another method for combining trees into a graph while retaining all of the original information from the source trees is the Tree Alignment Graph (TAG), an idea introduced by Stephen A. Smith, Joseph W. Brown and Cody E. Hinchliff (2013. Analyzing and synthesizing phylogenies using tree alignment graphs. PLoS Computational Biology 9: e1003223).
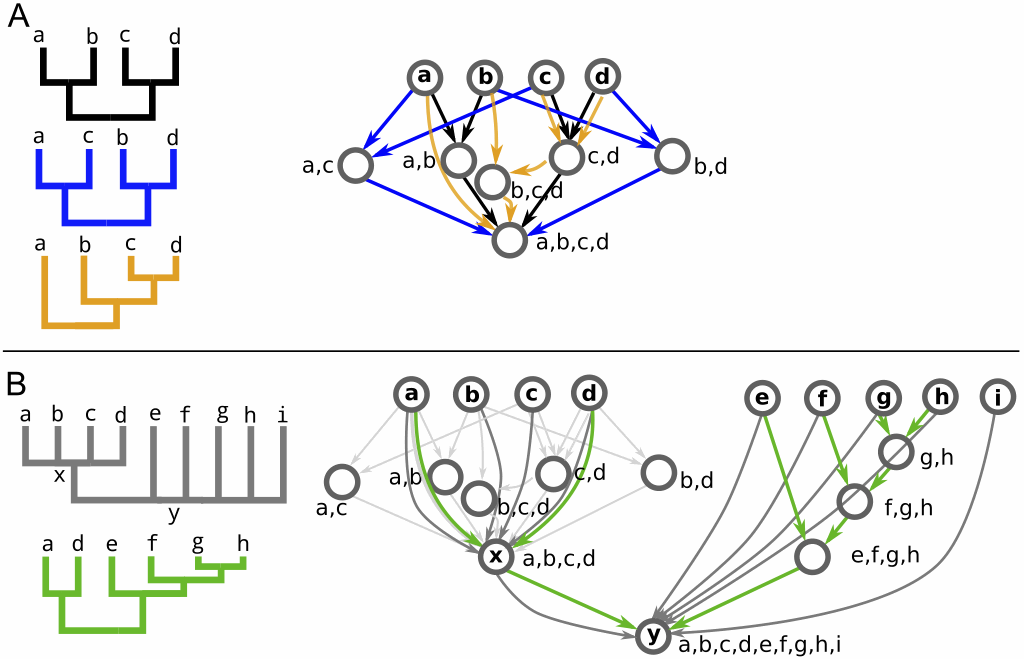
The authors note:
These methods address the problem of identifying common nodes and edges across sets of phylogenetic trees and constructing a data structure that efficiently contains this information while retaining original source information ... Mapping trees into a TAG exploits the fact that rooted phylogenetic trees are in fact a specific type of graph: they are directed, acyclic, and require that each node has, at most, one parent. By relaxing these requirements, we can combine multiple trees into a common graph, while minimizing changes to the semantic interpretations of nodes and edges in the trees. Because they contain nodes and edges directly analogous to those from their source trees, TAGs have the desirable quality of retaining the full identifiability of the original source trees they contain. Additionally, because they are not restricted to the bifurcating model of evolution, TAGs may represent conflict among source trees as reticulations in the graph.The basic principal is illustrated in the first figure (about). Internal nodes represent collections of terminal nodes, and arcs (directed edges) represent their relationships. Nodes and arcs are added to the growing TAG, each of which represents one relationship shown in one of the original trees. TAG A in the figure shows the result of combining the black, blue and orange trees, while TAG B shows the result of then adding the gray and green trees to TAG A (the arcs are colour-coded). The resulting TAG is thus a database of all of the original information, which can then be queried in any way to provide summaries of the data. In particular, standard network summaries can be used, such as node degree, which will highlight parts of the TAG with interesting characteristics.

The authors provide two empirical examples of applications. The one shown here involves 100 bootstrap trees for 640 species representing the majority of known lineages from the Angiosperm Tree of Life dataset (chloroplast, mitochondrial, and ribosomal data). The TAG is shown lightly in the background. Superimposed on this, the nodes are coloured to represent the effective number of parent nodes, and their size represents node bootstrap support. Highly supported nodes with a low number of effective parents (large blue nodes) are frequently recovered and confidently placed in the source trees, while highly supported nodes with a low number of effective parents (large and pink or orange) are frequently resolved in the source trees but their placement varies among bootstrap replicates. So, the three largest problem areas as illustrated in the TAG correspond to the Malpighiales, Lamiales and Ericales.
For comparison, a NeighborNet analysis of the same data is shown in the blog post When is there support for a large phylogeny? This simply shows an unresolved blob.
Posted by in Cloudogram, DAG
Phylogenetic networks and "evolutionary networks"
Complex networks are found in all parts of biology, graphically representing biological patterns and, if they are directed networks, also their causal processes. Directed networks are currently used to model various aspects of biological systems, such as gene regulation, protein interactions, metabolic pathways, ecological interactions, and evolutionary histories.
Two types of networks can be distinguished, and this distinction seems to me to be very important. Most networks are what might be called observed networks, in the sense that the nodes and edges represent empirical observations. For example, a food web consists of nodes representing animals with connecting edges representing who eats whom. Similarly, in a gene regulation network the genes (nodes) are connected by edges showing which genes affect the functioning of which other genes. In all cases, the presence of the nodes and edges in the graph is based on experimental data. These are collectively called interaction networks or regulation networks.
However, when studying historical patterns and processes not all of the nodes and edges can be observed. So, instead, they are inferred as part of the data-analysis procedure. That is, we infer the patterns as well as the processes; and we can call these inferred networks. In this case, the empirical data may consist solely of the leaf nodes, and we infer the other nodes plus all of the edges. For example, every person has two parents, and even if we do not observe those parents we can infer their existence with confidence, as we also can for the grandparents, and so on back through time with a continuous series of ancestors. Alternatively, we may also observe some of the internal nodes of the network, such as when we do record the parents and grandparents because they are contemporaneous (ie. their generations overlap). This type of pattern can be represented as a genealogical network, when referring to individual organisms, or a phylogenetic network when referring to groups (populations, species, or larger taxonomic groups).
What, then, are the things often referred to as "evolutionary networks" but which are clearly not phylogenetic networks? They are of the first type, the interaction networks. In an evolutionary network the observed nodes are directly connected to each other to represent some aspect of evolution. This aspect may have some component of phylogeny to it, but there is more to the study of evolution than solely phylogenetic history.
For example, directed LGT (dLGT) networks connect nodes representing contemporary organisms with edges that represent inferred lateral gene transfer. That is, the evolutionary networks show gene sharing. This is obviously related to the phylogeny of the organisms, but the network does not display the phylogeny itself. This first example (from Ovidiu Popa, Einat Hazkani-Covo, Giddy Landan, William Martin, Tal Dagan. 2011. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Research 21: 599-609) shows "32,028 polarized lateral recipient–donor protein-coding gene transfer events" inferred from "the completely sequenced genomes of 657 prokaryote species".

The concept of a gene-sharing network as an evolutionary network has also been applied to viruses and their relatives, for example, as shown by this next diagram (from Natalya Yutin, Didier Raoult, Eugene V Koonin. 2013. Virophages, polintons, and transpovirons: a complex evolutionary network of diverse selfish genetic elements with different reproduction strategies. Virology Journal 10: 158).

The question, then, is what to make of diagrams that combine both a phylogenetic tree and this type of evolutionary network, such as is done in the Minimal Lateral Network. This next example is from linguistics rather than biology (from Johann-Mattis List, Shijulal Nelson-Sathi, Hans Geisler, William Martin. 2013. Networks of lexical borrowing and lateral gene transfer in language and genome evolution. Bioessays 36: 141-150), and it superimposes the sharing network and the phylogenetic tree. (For a discussion in the context of LGT, see also Tal Dagan. 2011. Phylogenomic networks. Trends in Microbiology 19: 483-491).

In this diagram, the tree explicitly represents the phylogenetic history of the languages while the evolutionary network represents possible borrowings of words, with thicker lines representing more borrowed words. Clearly, the network also contains phylogenetic information of some sort. For example, the connection of the root of the Romance languages to English reflects the conquest of Britain by the French-speaking Normans, which modified the Old-German heritage of Old English. However, the diagram as a whole is a hybrid, rather than being a coherent phylogenetic network in the simplest sense (ie. a reticulation network).
To see this clearly, note that the phylogenetic tree is not fully resolved and that the evolutionary network does suggest possible resolutions for several of polychotomies, such as the relationship of Armenian and Greek, the relationship of Albanian to the Romance languages, and the relationship of the Gaelic languages to the Romance languages. So, in some cases the evolutionary network helps resolve the phylogenetic tree rather than forming a reticulating network.
It would be possible to derive a phylogenetic network from this minimal lateral network, but as it stands it is a combination of a phylogenetic tree and a so-called evolutionary network.
Posted by in DAG, Evolutionary network, phylogeny
Monophyletic groups in networks
I have noted before that taxonomic groups that are represented in any tree-like parts of a phylogeny can be considered to be monophyletic, but those that consist of hybrids cannot, unless we hypothesize a single hybrid origin for each group (How should we treat hybrids in a taxonomic scheme?). This issue arises from the concept that monophyletic groups must share an exclusive Most Recent Common Ancestor (MRCA), and this concept is not straightforward for a network compared to a tree.
This topic has been tackled mathematically a couple of times (see Huson and Rupp 2008; Fischer and Huson 2010), resulting in the recognition that for a network there are three main types of MRCA: conservative MRCA (or stable MRCA), Lowest Common Ancestor (or minimal common ancestor), and Fuzzy MRCA (see Networks and most recent common ancestors). These have definitions based on the Least Lower Bound and Greatest Lower Bound of mathematical lattices.
Unfortunately, there has been very little discussion of the topic in the biological literature. However, recently Wheeler (2013) has made a start. There is no reference to the mathematical work on MRCAs, but he considers what to do about the concepts of monophyly, paraphyly and polyphyly with respect to networks.
Basically, he suggests three new types of phyletic group: periphyletic, epiphyletic, and anaphyletic. He provides algorithmic definitions of these groups, relating them to the previous algorithmic definitions of monophyly, paraphyly and polyphyly. These new types concern groups that are monophyletic on a tree, but have additional gains or losses of members from network edges — that is, they lie somewhere between monophyletic and paraphyletic.
For example, an epiphyletic group would be one that is otherwise monophyletic but also contains one or more hybrids that have one of their parents from outside the group, while a periphyletic group would be monophyletic but has contributed as a parent to at least one hybrid outside the group. An anaphyletic group would have done both of these things. For clarification, Wheeler provides the following empirical example, based on Indo-European languages (where English is recognized as a "hybrid" of Germanic and Romance languages).
 |
| Reproduced from Wheeler (2013). |
In terms of MRCA, it seems to me that all three new group types use the Lowest Common Ancestor model, which is the shared ancestor that is furthest from the root along any path (ie. the LCA is not an ancestor of any other common ancestor of the taxa concerned). However, this is only clear when we consider hybrids, in which the two (or more) parents contribute equally to the hybrid offspring. When dealing with introgression or horizontal gene transfer, where the parentage is unequal, then we approach the Fuzzy MRCA model, in which only a specified proportion of the paths (representing some proportion of the genomes) needs to be accommodated by the MRCA, thus keeping the MRCA close to the main collection of descendants.
What is not yet clear is whether we would want to recognize any of these new group types in a taxonomic scheme. I guess that this is something that the PhyloCode will have to think about, since it is based strictly on clades (although they are allowed to overlap).
References
Fischer J, Huson DH (2010) New common ancestor problems in trees and directed acyclic graphs. Information Processing Letters 110: 331–335.
Huson DH, Rupp R (2008) Summarizing multiple gene trees using cluster networks. Lecture Notes in Bioinformatics 5251: 296–305.
Wheeler WC (2013) Phyletic groups on networks. Cladistics (online early).
Posted by in DAG, Directed graph, Hybridization network, MRCA
Posted by in DAG, HGT network, Hybridization network, inference, phylogeny
Networks and most recent common ancestors
The interpretation of an evolutionary network is confounded by the fact that descendants of reticulation nodes have complex ancestry. Therefore, the concept of a Most Recent Common Ancestor (MRCA) is not as straightforward as it is for a tree, as there may be multiple paths from any one descendant back to its ancestors. This creates several possible interpretations of what we might mean by a MRCA.
Figure 1 illustrates the calculation of the MRCA in a tree of five taxa (A-E), showing the MRCA of taxa C and D. We simply trace each of the descendant taxa backward along the branches towards the root, and the ancestral node where all of these traces first intersect is the MRCA of those taxa.
 |
| Figure 1. |
Figure 2 illustrates a more complex history, involving two hybridization events. The incoming branches to the reticulation nodes have arrows, for emphasis. The figure also recognizes several possible interpretations of the MRCA of taxa C and D (see Huson and Rupp 2008; Fischer and Huson 2010).
A conservative definition of the MRCA (or a stable MRCA) is the intersection of all paths from the descendants to the root, so that any reticulation pushes the MRCA back towards the root. In this example it pushes the MRCA all the way to the root. Alternatively, we could define the Lowest Common Ancestor (or the minimal common ancestor) as the shared ancestor that is furthest from the root along any path. That is, the LCA is not an ancestor of any other common ancestor of the taxa concerned.
 |
| Figure 2. |
In the mathematical terminology of lattices, which can have an algebraic or order theoretic definition, the Conservative MRCA is called the Least Lower Bound (LLB) and the LCA is called the Greatest Lower Bound (GLB).
We could also have a biological compromise between these two mathematical concepts and recognize a Fuzzy MRCA, in which only a specified proportion of the paths (representing some proportion of the genomes) needs to be accommodated by the MRCA, thus keeping the MRCA close to the main collection of descendants (Fischer and Huson 2010). In this example, the Fuzzy MRCA represents 75% of the genome of taxon C and 100% of the genome of taxon D. (The Conservative MRCA represents 100% for both taxa, by definition; and in this example the LCA represents 50% of the genome of taxon C and 100% of the genome of taxon D.)
 |
| Figure 3. |
However, neither the Fuzzy MRCA nor the LCA is necessarily unique, although the Conservative MRCA will always be unique. Figure 3 shows an example where there are two independent LCAs of taxa C and D. Neither of these LCAs is an ancestor of the other, as required by the definition, and so they are both equal candidates as LCA. Each one represents 50% of the genome for both taxa C and D.
In terms of a lattice, Figure 2 is called a lower semi-lattice (or meet semi-lattice), because every pair of nodes has only one GLB, whereas Figure 3 is not a semi-lattice, because at least one node pair has more than one GLB.
This leads to the biological question of how we are best to interpret the MRCA in situations such as that represented by Figure 3. This is a question that does not yet seem to have been addressed by biologists. Figure 3 does not represent an impossible evolutionary history, although it may be an unusual one because one lineage hybridizes with another lineage twice, presumably at different times.
The lack of a unique LCA is clearly problematic, as it almost defeats the purpose of the concept of a MRCA. It would certainly make life easier if we could restrict evolutionary networks to the class of lower semi-lattices.
An alternative is to restrict the MRCA concept to the Conservative MRCA. However, it is easy to imagine situations where this pushes the MRCA so far towards the root of the network as to be uninformative, especially in cases involving horizontal gene transfer, which can occur between widely separated evolutionary groups. If we insist that a eukaryote MRCA represent 100% of the genome, and we include non-nuclear genomes in the calculation, then the Conservative MRCA creates an extreme theoretical problem.
A Fuzzy MRCA may be the best compromise between these two extremes, although there are obvious practical issues for obtaining agreement on how much of the genome history is to be discounted from the MRCA.
References
Fischer J., Huson D.H. (2010) New common ancestor problems in trees and directed acyclic graphs. Information Processing Letters 110: 331–335.
Huson D.H., Rupp R. (2008) Summarizing multiple gene trees using cluster networks. Lecture Notes in Bioinformatics 5251: 296–305.
Posted by in DAG, Directed graph, MRCA
Are mathematical constraints biologically realistic?
Mathematicians and other computational scientists have produced their own definitions of phylogenetic networks, independently of biologists. For the evolutionary type of phylogenetic network, the definition usually looks something like this:
A phylogenetic network is a rooted, directed graph (consisting of nodes, plus edges that connect each parent node to its child nodes) such that:
(1) There is exactly one node having indegree 0, the root
- all other nodes have indegree 1 or 2
(2) All nodes with indegree 1 have outdegree 2 or 0
- nodes with outdegree 2 are tree nodes
- nodes with outdegree 0 are leaves, distinctly labelled
(3) The root has outdegree 2, and
(4) Nodes with indegree 2 have outdegree 1, called reticulation nodes.
An obvious question of interest is how (or whether?) this definition connects to what biologists have in mind when they use the term "phylogenetic network". Clearly, this definition places considerable restrictions on the networks that will be inferred by any mathematical algorithm, which in turn affects their use as models for biological inference.
The first thing to note is that unrooted networks are excluded, because the graph is directed. Thus, many (if not most) of the phylogenetic networks that have appeared in the literature are excluded from the discussion. Furthermore, a tree is considered to have all internal nodes with indegree 1 and outdegree 2 (i.e. no reticulation nodes), and we know this to be biologically unrealistic, in general. (Otherwise, this blog would be redundant!)
Biologically, the other parts of the definition imply:
One node of indegree 0
- the network has no previous ancestry that is to be inferred
Nodes with outdegree 0 are labelled
- observed (contemporary) taxa occur only at the leaves
All nodes with indegree 2 have outdegree 1
- reticulation and speciation cannot occur simultaneously
No nodes with indegree >2
- reticulation events cannot involve input from more than 2 parents simultaneously
No nodes with outdegree >2
- speciation involves only two children at a time.
These do not appear to be onerous biological restrictions. Indeed, he first two have been standard characteristics of tree-building for several decades. The other three are also logical extensions of the restrictions that have previously been placed on trees. However, phylogenetic history is unlikely to have been as simple as implied by these features. Thus, biologists will need to keep a careful eye on whether the simplifications are affecting the networks inferred for their particular group of organisms.
Other restrictions
In addition to the restrictions created by the definition, other topological restrictions have been used to make the mathematical inference algorithms computationally tractable. Thus, only certain sub-families of possible networks are considered by most of the computer programs. These include:
- tree-child network, tree-sibling network
- level-k network, galled tree
- binary input trees for hybridization and HGT networks
- binary characters for recombination networks.
Tree-child, Tree-sibling
In a tree-child network, every internal node has at least one child node that is a tree node
- ie. a reticulation event cannot be followed immediately by another reticulation event
In a tree-sibling network, every reticulation node has at least one sibling node that is a tree node
- ie. a parent cannot be directly involved in two separate reticulation events
Note that every tree-child network will also be a tree-sibling network, but not vice versa.
Algorithmically, these two restrictions may involve the addition of extra tree nodes to an inferred network, in order to satisfy the restrictions. Biologically, the question is whether real networks are this simple. Arenas et al. (2008) simulated data under the coalescent with recombination, and found that even at small recombination rates most of the networks produced were already more complex than a tree-sibling network. On the other hand, Arenas et al. (2010) analyzed real population-level data from the PopSet and Polymorphix databases using the TCS program, and found that >98% the resulting networks could be characterized as tree-sibling. So, there is cause for optimism, in the sense that the "optimum" networks algorithmically are not necessarily complex, at least for closely related organisms (ie. within species).
Level-k network, Galled tree
A network has level k if each tangled part of the network (ie. each biconnected component) contains at most k reticulation nodes (see this previous post). This is a generalization of the older notion of galled trees, in which reticulation cycles do not overlap (ie. do not share edges or nodes), as galled trees are level-1 networks. Level-k networks can also be seen as a generalization of networks with k reticulation nodes, although there may be a difference between a network with minimum level and one with a minimum number of reticulations.
Algorithmically, these restrictions have been used to guide the search for (or choice of) the "optimal" inferred network. Biologically, these notions do not seem to have been investigated, but basically they restrict how complex inferred reticulation histories can be. In particular, they restrict the complexity of any given subset of each network. It has been noted that optimizing k can easily lead to networks that look biologically unrealistic (Huson et al. 2011).
Binary input
The requirements for binary input trees and binary characters are restrictions that have been applied in the past, because they greatly reduce the complexity of the input to the network algorithms, but they are now being relaxed. Effectively, the restrictions are to fully dichotomous trees and SNP characters. These are not unusual restrictions in evolutionary analysis, but they are obviously unrealistic.
As I noted in an earlier blog post, non-binary data often reflect uncertainty in the input, rather than a strictly bifurcating history, and this is not taken into account in the network inference if the input is restricted to a binary state. In particular, it may be unnecessarily hard to construct a network (because not all of the data signals relate to reticulation), and the resulting networks may have far too many reticulation nodes.
Conclusion
It is still an open question about the extent to which we can use these topologically restricted families of mathematical networks as a basis for reconstructing biological histories. Clearly, much more work is needed to understand the connections between the mathematical restrictions and the requirements of biological modelling.
References
M. Arenas, M. Patricio, D. Posada, G. Valiente (2010) Characterization of phylogenetic networks with NetTest. BMC Bioinformatics 11: 268.
M. Arenas, G. Valiente, D. Posada (2008) Characterization of reticulate networks based on the coalescent with recombination. Molecular Biology and Evolution 25: 2517-2520.
D. H. Huson, R. Rupp, C. Scornavacca (2011) Phylogenetic Networks: Concepts, Algorithms and Applications. Cambridge University Press.
Posted by in DAG, galled trees, inference, Level-k network, optimization criteria