I will be talking at the Festival of Genomics on Wednesday 24 January about Identifying virulence and antimicrobial resistance genes in bacterial using genome-wide association studies. You can preview my talk here.
Category Archives: Staphylococcus aureus
New paper: Antimicrobial resistance determinants are associated with Staphylococcus aureus bacteraemia and adaptation to the healthcare environment
Staphylococcus aureus is a leading cause of infectious disease deaths in all countries, with bloodstream infection leading to sepsis a major concern. This new study, published in November in Microbial Genomics, reports genes and genetic variants in Staph. aureus associated severe disease vs asymptomatic carriage, and healthcare vs community carriage.
Our genome-wide association study of 2000 bacterial genomes showed that antibiotic resistance in Staph. aureus is associated with severe disease and the hospital environment:
- A mutation conferring trimethoprim resistance (dfrB F99Y) and the presence of a gene conferring methicillin resistance (mecA) were both associated with bloodstream infection vs asymptomatic nose carriage.
- Separately, we demonstrated that a mutation conferring fluoroquinolone resistance (gyrA L84S) and variation in a gene involved in resistance to multiple antibiotics (prsA) were preferentially associated with healthcare-associated carriage vs community-acquired carriage.
The implication – that antibiotic resistance genes may provide survival advantages which mechanistically contribute to the development of disease – is important in the face of the continued global rise of antibiotic resistance.
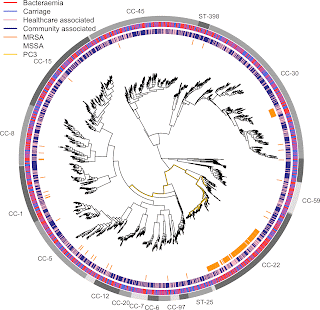
We were also able to shed light on a controversy as to whether different strains of Staph. aureus differ in their propensity to cause severe disease. Interest in this question dates back decades in the literature, and contradictory studies, often based on modest sample sizes, have reached different conclusions. Our comparatively large study, using a whole-genome method that we previously published in Nature Microbiology, found that all strains of Staph. aureus are equally likely to cause severe disease vs asymptomatic carriage.
New paper: PVL toxin associated with pyomyositis
In a new collaborative study published this week in eLife, we report a strong association between Staphylococcus aureus that carry the PVL toxin and pyomyositis, a muscle infection often afflicting children in the tropics.
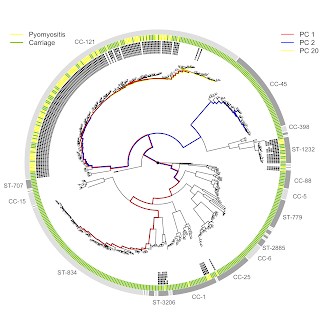 Catrin Moore and colleagues at the Angkor Children's Hospital in Siem Reap, Cambodia, spent more than a decade collecting S. aureus bacteria from pyomyositis infections in young children, and built a comparable control group of S. aureus carried asymptomatically in children of similar age and location.
Catrin Moore and colleagues at the Angkor Children's Hospital in Siem Reap, Cambodia, spent more than a decade collecting S. aureus bacteria from pyomyositis infections in young children, and built a comparable control group of S. aureus carried asymptomatically in children of similar age and location.
When Bernadette Young in our group compared the genomes of cases and controls using statistical tools we developed, she found some strong signals:
 Catrin Moore and colleagues at the Angkor Children's Hospital in Siem Reap, Cambodia, spent more than a decade collecting S. aureus bacteria from pyomyositis infections in young children, and built a comparable control group of S. aureus carried asymptomatically in children of similar age and location.
Catrin Moore and colleagues at the Angkor Children's Hospital in Siem Reap, Cambodia, spent more than a decade collecting S. aureus bacteria from pyomyositis infections in young children, and built a comparable control group of S. aureus carried asymptomatically in children of similar age and location.When Bernadette Young in our group compared the genomes of cases and controls using statistical tools we developed, she found some strong signals:
- Most, but not all, pyomyositis was caused by the CC-121 strain, common in Cambodia.
- The association with CC-121 was driven by the PVL toxin which it carries.
The ability to pinpoint the association to PVL came about because (i) a sub-group of CC-121 that lacked PVL caused no pyomyositis and (ii) pyomyositis-causing S. aureus from backgrounds that rarely caused pyomyositis were unusual in also possessing PVL.
The strength of the PVL-pyomyositis association was extraordinarily strong, so strong that PVL appeared all-but necessary for disease. Moreover, disease appeared to be monogenic, with no other genes involved elsewhere in the bacterial genome. To discover an apparently monogenic disease mechanism for a common disease is very unusual nowadays.
The discovery has immediate practical implications because it draws parallels between pyomyositis and toxin-driven bacterial diseases like tetanus and diphtheria that have proven amenable to immunization. The fact that anti-PVL vaccines have already been developed in other contexts offers hope for the future treatment of this debilitating tropical infection.
Our study throws much-needed light on a subject that has been the subject of heated debate over previous years. Many bacterial toxins, PVL included, have been implicated in diverse S. aureus disease manifestations, often without sound evidence. Because PVL is known to contribute to angry, pus-filled skin infections, and has been observed in bacteria causing rare and severe S. aureus infections, some authors have implicated it in dangerous diseases including necrotizing pneumonia, septic arthritis and pyomyositis, but detailed meta-analyses have dismissed these claims as not substantiated. Our GWAS approach offers unprecedented robustness over previous generations of candidate gene studies by accounting for bacterial genetic variation across the entire genome.
If you are interested, please take a closer look at the paper.
Bacterial Doubling Times in the Wild
How fast do bacteria grow outside the laboratory? This simple question is very difficult to address directly, because it is near-impossible to track a lineage of bacterial cells, ancestor-to-decendant, inside an infected patient or through a river. Now in new work published in Proceedings B, Beth Gibson, Ed Feil, Adam Eyre-Walker and I exploit genome sequencing to try to get a handle on the problem indirectly.
We have done it by comparing two known quantities and taking the ratio: the rate at which DNA mutates in bacteria per year, and the rate it mutates per replication. This tells us in theory how many replications there are per year.
The mutation rate per replication has long been studied in the laboratory, and is around once per billion letters. Meanwhile, the recent avalanche of genomic data has allowed microbiologists to quantify the rate at which bacteria evolve over short time scales such as a year, including during outbreaks and even within individual infected patients. Most bugs mutate about once per million letters per year, with ten-fold variation above and below this not uncommon among different species.
For five species both these quantities exist. The fastest bug we looked at causes cholera and we estimate it doubles once every hour on average (give or take 30 minutes). The slowest was Salmonella, which we estimate doubles once a day on average (give or take 8 hours). In between were Staph. aureus and Pseudomonas at about two hours each, and E. coli at 15 hours. These are average over the very diverse and often hostile conditions that a bacterial cell may find itself in during the course of its natural lifecycle. To find out more about the work, please check out the paper.
We have done it by comparing two known quantities and taking the ratio: the rate at which DNA mutates in bacteria per year, and the rate it mutates per replication. This tells us in theory how many replications there are per year.
The mutation rate per replication has long been studied in the laboratory, and is around once per billion letters. Meanwhile, the recent avalanche of genomic data has allowed microbiologists to quantify the rate at which bacteria evolve over short time scales such as a year, including during outbreaks and even within individual infected patients. Most bugs mutate about once per million letters per year, with ten-fold variation above and below this not uncommon among different species.
For five species both these quantities exist. The fastest bug we looked at causes cholera and we estimate it doubles once every hour on average (give or take 30 minutes). The slowest was Salmonella, which we estimate doubles once a day on average (give or take 8 hours). In between were Staph. aureus and Pseudomonas at about two hours each, and E. coli at 15 hours. These are average over the very diverse and often hostile conditions that a bacterial cell may find itself in during the course of its natural lifecycle. To find out more about the work, please check out the paper.
Postdoc positions in Data Science and Molecular Microbiology
These positions are now closed
As part of the move to the Big Data Institute, two new postdoctoral positions funded by the Robertson Foundation are available in Data Science and Molecular Microbiology.
The BDI is a new interdisciplinary research centre aiming to develop, evaluate and deploy efficient methods for acquiring and analysing biomedical data at scale and for exploiting the opportunities arising from such studies. The BDI is a joint venture between the renowned Nuffield Department of Population Health (NDPH) and NDM.
The Data Scientist role, split between the BDI and London, will be part of a team developing systems for continuous record linkage between Public Health England and other population health records. The aims are to design record linkage algorithms, manage front ends for viewing the data source, and analyse and interpret results. We're looking for a graduate or equivalent experience in computer science, data science, statistics, or any other relevant subject with a strong quantitative component. Knowledge of databases like SQL and computer programming are needed.
The Molecular Microbiology role, based mainly at the John Radcliffe Hospital Microbiology Department, will be part of a team researching Staphylococcus aureus infection using RNA sequencing, genome wide association studies, and biochemical and immunological assays of bacterial behaviour. The aims include designing microbiological protocols, researching bacterial molecular genetics and data analysis. We're looking for a PhD or equivalent experience in a relevant subject such as microbiology, immunology, genetics or biochemistry. Experience designing protocols and basic microbiological and immunological skills are required.
The deadline for the posts is Noon on 6 June 2018. Both are one year positions. For more details or to apply click here for the Data Scientist role and here for the Molecular Microbiologist role.
As part of the move to the Big Data Institute, two new postdoctoral positions funded by the Robertson Foundation are available in Data Science and Molecular Microbiology.
The BDI is a new interdisciplinary research centre aiming to develop, evaluate and deploy efficient methods for acquiring and analysing biomedical data at scale and for exploiting the opportunities arising from such studies. The BDI is a joint venture between the renowned Nuffield Department of Population Health (NDPH) and NDM.
The Data Scientist role, split between the BDI and London, will be part of a team developing systems for continuous record linkage between Public Health England and other population health records. The aims are to design record linkage algorithms, manage front ends for viewing the data source, and analyse and interpret results. We're looking for a graduate or equivalent experience in computer science, data science, statistics, or any other relevant subject with a strong quantitative component. Knowledge of databases like SQL and computer programming are needed.
The Molecular Microbiology role, based mainly at the John Radcliffe Hospital Microbiology Department, will be part of a team researching Staphylococcus aureus infection using RNA sequencing, genome wide association studies, and biochemical and immunological assays of bacterial behaviour. The aims include designing microbiological protocols, researching bacterial molecular genetics and data analysis. We're looking for a PhD or equivalent experience in a relevant subject such as microbiology, immunology, genetics or biochemistry. Experience designing protocols and basic microbiological and immunological skills are required.
The deadline for the posts is Noon on 6 June 2018. Both are one year positions. For more details or to apply click here for the Data Scientist role and here for the Molecular Microbiologist role.
New paper: Severe infections emerge from commensal bacteria by adaptive evolution

This study shows that the emergence of life-threatening infections of the major pathogen Staphylococcus aureus from bacteria colonizing the nose is associated with repeatable adaptive evolution inside the human body.
First author Bernadette Young has summarized the paper's findings on the Modernising Medical Microbiology blog.
Posted by in Adaptation, Bernadette Young, eLife, Evolution, Staphylococcus aureus
The Rsp virulence regulator: new review in Trends in Microbiology
In the September issue of Trends in Microbiology, Mark Smeltzer casts the spotlight on the story of rsp, a virulence regulator in Staphylococcus aureus that evolves within infected patients and may play a role in disease.
The new review covers recent work on the rsp gene including a series papers that my collaborators and my group have contributed:
Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation.
Das, S., Lindemann, C., Young, B. C., Muller, J., Österreich, B., Ternette, N., Winkler, A.-C., Paprotka, K., Reinhardt, R., Förstner, K. U., Allen, E., Flaxman, A., Yamaguchi, Y., Rollier, C. S., Van Diemen, P., Blättner, S., Remmele, C. W., Selle, M., Dittrich, M., Müller, T., Vogel, J., Ohlsen, K., Crook, D., Massey, R., Wilson, D. J., Rudel, T., Wyllie, D. H., and M. J. Fraunholz (2016)
Proceedings of the National Academy of Sciences USA 113: E3101–E3110. (abstract pdf)
Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus.
Laabei, M., Uhlemann, A.-C., Lowy, F. D., Austin, E. D., Yokoyama, M., Ouadi, K., Feil, E., Thorpe, H. A., Williams, B., Perkins, M., Peacock, S. J., Clarke, S. R., Dordel, J., Holden, M., Votintseva, A. A., Bowden, R., Crook, D. W., Young, B. C., Wilson, D. J., Recker, M. and R. C. Massey (2015)
PLoS Biology 13: e1002229. (abstract pdf)
Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease.
Young, B. C., Golubchik, T., Batty, E. M., Fung, R., Larner-Svennson, H., Votintseva, A., Miller, R. R., Godwin, H., Knox, K., Everitt, R. G., Iqbal, Z., Rimmer, A. J., Cule, M., Ip C. L. C., Didelot, X., Harding, R. M., Donnelly, P. J., Peto, T. E., Crook, D. W., Bowden, R. and D. J. Wilson (2012)
Proceedings of the National Academy of Sciences USA 109: 4550-4555. (abstract pdf F1000)
The new review covers recent work on the rsp gene including a series papers that my collaborators and my group have contributed:
Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation.
Das, S., Lindemann, C., Young, B. C., Muller, J., Österreich, B., Ternette, N., Winkler, A.-C., Paprotka, K., Reinhardt, R., Förstner, K. U., Allen, E., Flaxman, A., Yamaguchi, Y., Rollier, C. S., Van Diemen, P., Blättner, S., Remmele, C. W., Selle, M., Dittrich, M., Müller, T., Vogel, J., Ohlsen, K., Crook, D., Massey, R., Wilson, D. J., Rudel, T., Wyllie, D. H., and M. J. Fraunholz (2016)
Proceedings of the National Academy of Sciences USA 113: E3101–E3110. (abstract pdf)
Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus.
Laabei, M., Uhlemann, A.-C., Lowy, F. D., Austin, E. D., Yokoyama, M., Ouadi, K., Feil, E., Thorpe, H. A., Williams, B., Perkins, M., Peacock, S. J., Clarke, S. R., Dordel, J., Holden, M., Votintseva, A. A., Bowden, R., Crook, D. W., Young, B. C., Wilson, D. J., Recker, M. and R. C. Massey (2015)
PLoS Biology 13: e1002229. (abstract pdf)
Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease.
Young, B. C., Golubchik, T., Batty, E. M., Fung, R., Larner-Svennson, H., Votintseva, A., Miller, R. R., Godwin, H., Knox, K., Everitt, R. G., Iqbal, Z., Rimmer, A. J., Cule, M., Ip C. L. C., Didelot, X., Harding, R. M., Donnelly, P. J., Peto, T. E., Crook, D. W., Bowden, R. and D. J. Wilson (2012)
Proceedings of the National Academy of Sciences USA 109: 4550-4555. (abstract pdf F1000)
New paper: How low-toxic Staph. aureus mutants cause severe infections

reduce toxicity while maintaining the ability to survive, proliferate and cause infection within the human body.
In previous work, we have found that Staph. aureus evolves by mutation within the body quickly enough to influence the progression of disease, and that diversity generated by evolution in the body is a widespread phenomenon. In the case of one patient who we followed longitudinally for over a year, we identified that bacteria in the bloodstream differed from those in the nose by several mutations, of which a loss-of-function mutation in the rsp regulatory gene represented the most likely candidate for playing a possible role in causing severe infection.
We collaborated with Ruth Massey at Bath who discovered to our surprise that while rsp loss-of-function mutants do indeed show differences in toxicity - one of several traditional correlates of virulence readily measured in the laboratory - they showed reduced toxicity. Going further, Ruth and her collaborators showed that bloodstream infections in general show reduced toxicity compared to milder skin infections and asymptomatically carried nose populations, overturning previous views on the relationship between Staph. aureus toxicity and virulence.
Today's new paper offers a detailed dissection of rsp. Working with Claudia Lindemann and David Wyllie at the University of Oxford and Martin Fraunholz and collaborators at the University of Würzburg, we found that although rsp mutants show reduced toxicity, crucially they retain their capacity to survive, grow, spread through the body and cause abscesses. In other words, rsp uncouples toxicity from pathogenicity. This decoupling could be important for evading the immune system and establishing severe infections. To find out more, see the full paper.
Making the most of bacterial GWAS: new paper in Nature Microbiology
In a new paper published this week in Nature Microbiology, we report the performance of genome wide association studies (GWAS) in bacteria to identify causal mechanisms of antibiotic resistance in four major pathogens, and introduce a new method, bugwas, to make the most of bacterial GWAS for traits under less strong selection.
As explained by Sarah Earle, joint first author with Jessie Wu and Jane Charlesworth, the problem with GWAS in bacteria is strong population structure and the consequent strong coinheritance of genetic variants throughout the genome. This phenomenon - known as genome-wide linkage disequilibrium (LD) - comes about because exchange of genes is relatively infrequent in bacteria, which reproduce clonally, compared to organisms that exchange genes every generation through sexual reproduction.
Genome-wide LD makes it difficult for GWAS to distinguish variants that causally influence a trait from other, coinherited variants that have no direct effect on the trait.
In the case of antibiotic resistance - a trait of high importance to human health - bacteria are under extraordinary selection pressures because resistance is a matter of life and death, to them as well as their human host. This helps overcome coinheritance and pinpoint causal variants because antibiotic usage selects for the independent evolution of the same resistance-causing variants in different genetic backgrounds.
Consequently, bacterial GWAS works very efficiently for antibiotic resistance: the variants most significantly associated with antibiotic resistance in 26 out of the 27 GWAS we performed were genuine resistance-conferring mutations. In the 27th we uncovered a putative novel mechanism of resistance to cefazolin in E. coli. These results for 17 antibiotics (ampicillin, cefazolin, cefuroxime, ceftriaxone, ciprofloxacin, erythromycin, ethambutol, fusidic acid, gentamicin, isoniazid, penicillin, pyrazinamide, methicillin, rifampicin, tetracycline, tobramycin and trimethoprim) across four species (E. coli, K. pneumoniae, M. tuberculosis and S. aureus) build on earlier work investigating beta-lactam resistance in S. pneumoniae, and convincingly demonstrate the potential for bacterial GWAS to discover new genes underlying important traits under strong selection.
What about traits under less strong selection, which probably includes pretty much every other bacterial trait? We show in this context that coinheritance poses a major challenge, based on detailed simulations. Often it may not be possible to use GWAS to pinpoint individual variants responsible for different traits because they are coinherited with - possibly many - other uninvolved variants.
But all is not lost. We show that even when individual locus-level effects cannot be pinpointed, there is often excellent power to characterize lineage-level differences in phenotype between strains. This is helpful for multiple reasons: (1) we often conceptualize trait variability in bacteria at the level of strain-to-strain differences (2) these differences can be highly predictive (3) we can prioritize variants for functional follow-up based on their contribution to strain-level differences.
These concepts represent a substantial departure from regular GWAS. In the human setting for instance, lineage-level differences are usually discarded as uninteresting or artefactual, and variants are almost always prioritized based on statistical evidence for involvement over-and-above any contribution to lineage-level differences. In the bacterial setting, we are forced to depart from these conventions because a large proportion of all genetic variation is strongly strain-stratified. To find out more, see the paper and try our methods.
As explained by Sarah Earle, joint first author with Jessie Wu and Jane Charlesworth, the problem with GWAS in bacteria is strong population structure and the consequent strong coinheritance of genetic variants throughout the genome. This phenomenon - known as genome-wide linkage disequilibrium (LD) - comes about because exchange of genes is relatively infrequent in bacteria, which reproduce clonally, compared to organisms that exchange genes every generation through sexual reproduction.
Genome-wide LD makes it difficult for GWAS to distinguish variants that causally influence a trait from other, coinherited variants that have no direct effect on the trait.
In the case of antibiotic resistance - a trait of high importance to human health - bacteria are under extraordinary selection pressures because resistance is a matter of life and death, to them as well as their human host. This helps overcome coinheritance and pinpoint causal variants because antibiotic usage selects for the independent evolution of the same resistance-causing variants in different genetic backgrounds.
Consequently, bacterial GWAS works very efficiently for antibiotic resistance: the variants most significantly associated with antibiotic resistance in 26 out of the 27 GWAS we performed were genuine resistance-conferring mutations. In the 27th we uncovered a putative novel mechanism of resistance to cefazolin in E. coli. These results for 17 antibiotics (ampicillin, cefazolin, cefuroxime, ceftriaxone, ciprofloxacin, erythromycin, ethambutol, fusidic acid, gentamicin, isoniazid, penicillin, pyrazinamide, methicillin, rifampicin, tetracycline, tobramycin and trimethoprim) across four species (E. coli, K. pneumoniae, M. tuberculosis and S. aureus) build on earlier work investigating beta-lactam resistance in S. pneumoniae, and convincingly demonstrate the potential for bacterial GWAS to discover new genes underlying important traits under strong selection.
What about traits under less strong selection, which probably includes pretty much every other bacterial trait? We show in this context that coinheritance poses a major challenge, based on detailed simulations. Often it may not be possible to use GWAS to pinpoint individual variants responsible for different traits because they are coinherited with - possibly many - other uninvolved variants.
But all is not lost. We show that even when individual locus-level effects cannot be pinpointed, there is often excellent power to characterize lineage-level differences in phenotype between strains. This is helpful for multiple reasons: (1) we often conceptualize trait variability in bacteria at the level of strain-to-strain differences (2) these differences can be highly predictive (3) we can prioritize variants for functional follow-up based on their contribution to strain-level differences.
These concepts represent a substantial departure from regular GWAS. In the human setting for instance, lineage-level differences are usually discarded as uninteresting or artefactual, and variants are almost always prioritized based on statistical evidence for involvement over-and-above any contribution to lineage-level differences. In the bacterial setting, we are forced to depart from these conventions because a large proportion of all genetic variation is strongly strain-stratified. To find out more, see the paper and try our methods.
PLoS Biology: Staphylococcus aureus invading the blood are less toxic
 |
| Toxicity in nose, blood and skin bacteria. |
The notion that isolates responsible for serious human infection are less toxic challenges some long-held beliefs about the mechanism of disease in Staphylococcus aureus infections. Most models of disease assume a straightforward relationship between increased toxicity and greater virulence - the propensity to cause, or severity of, disease.
To test her observation, Ruth collaborated with groups from New York and Cambridge to investigate whether the pattern observed in one patient held more generally across 134 Staphylococcus aureus belonging to the notorious USA300 strain. It did.
Curiously, bacteria isolated from the skin and from superficial infections were equally toxic to nose bacteria. These findings raise new questions about the role of toxicity in colonization, transmission and serious infections of Staphylococcus aureus. One possibility that we wish to investigate further is whether toxicity might be required for the usual transmission of Staphylococcus aureus populations in the nose, skin or superficial infections (such as impetigo), whereas loss of toxicity may promote transition to deep tissue and bloodstream infections by evading immune defences.
Posted by in plos biology, PNAS, Ruth Massey, Staphylococcus aureus, Virulogenomics