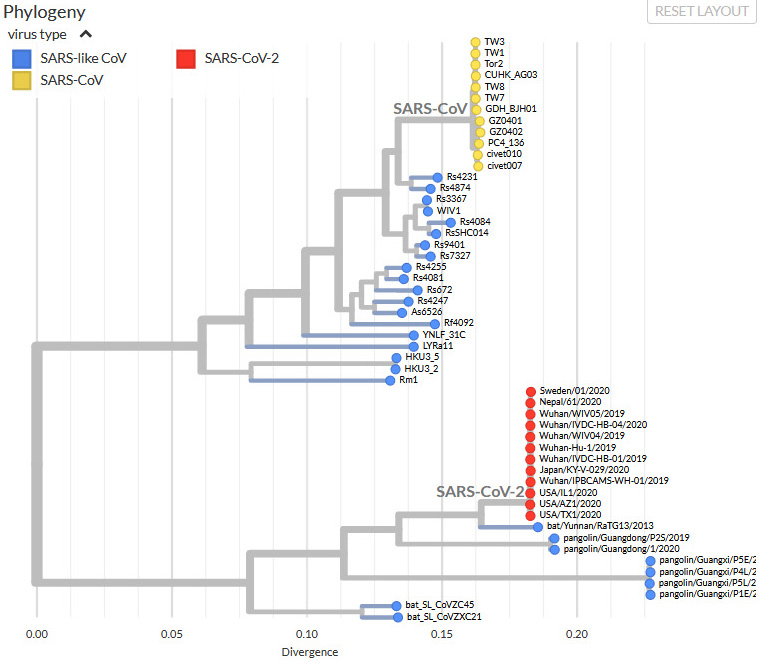
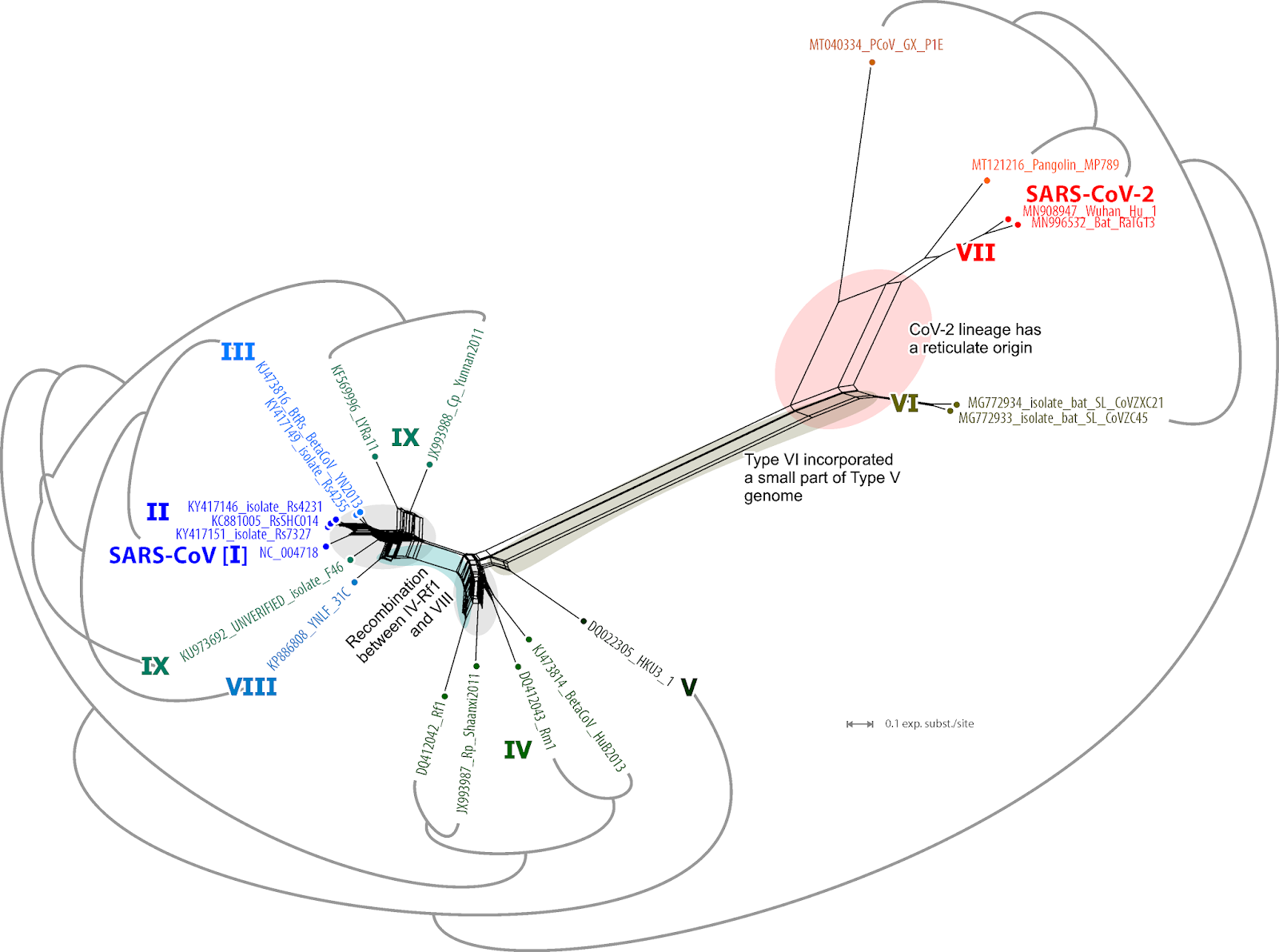
I have written a few blog posts recently about the current Covid-19 pandemic, caused by the arrival of the SARS-CoV-2 virus in our lives. This interests me as a biologist with some background in the study of pathogens (disease-causing organisms).
There have been two extreme responses to the current pandemic. There are all sorts of variants in between, of course, but I will start by characterizing the extremes, and then move on to some practical examples. The point here is that we need a reasoned response to this pandemic, based on the effect of the virus on people, and the make-up of the populations being affected. The current one-size-fits-all approach used by most governments is not going to work, long-term.
The future of having to live with the virus is becoming clearer. Actions can be individual, but they need to be co-ordinated, with each of the risk groups being treated appropriately. Even if you personally feel secure, those around you might experience risks very differently. An all-purpose set of mandated behaviors might work short-term, but we cannot continue to live that way. Behavior needs to make all risk groups feel safe at all times, by being targeted appropriately.
BehaviorsAt one extreme, people are trying to hide from the virus. By this, I mean that they are trying to keep away from it. Obviously, many people are doing this individually, but whole countries have also been trying to do it, notably Australia and New Zealand, which are geographically isolated by virtue of being islands. At the other extreme, people are trying to "crush" the virus, like they are playing poker against some weak opponent.
The problem with the first extreme is that you can never come out of hiding, because the virus does not go away, it just sits there (like viruses do) until you finally come past, and then it will get you, after all. This is what the so-called Second Wave of infections is currently showing us. The First Wave of infections occurs because people do not know about the pathogen, and therefore catch it inadvertently. In response to the rapid increase in case rates, people go into self-quarantine, trying to prevent themselves from encountering the virus. This works, but they eventually get tired of doing it, and they come back out again — and that is the Second Wave of infections. It is nothing new as far as the virus is concerned, it simply reflects changing human behavior (out, in, out again).
A prime example of the other extreme is expressed by this recent
New York Times article:
Here's how to crush the virus until vaccines arrive, or even the
Wall Street Journal:
The treatment that could crush Covid. You can't crush a pandemic, as we know from the seemingly endless series of previous pandemics in recorded history, and presumably many more of them before we learned to write. Naturally, Wikipedia has a
List of epidemics, for you to peruse.
However, at some stage, people are going to have to start treating the current pandemic like the influenza virus — a natural part of their environment, where they take standard precautions to minimize their risk. In response to the perennial threat of flu, old people take vaccines in winter, middle-aged people stay away from public transport during flu season, and young people simply get on with their lives (because a bit of flu will not kill them). These are rational responses, taken by people after evaluating the perceived risk of infection to themselves.
To do this for Covid-19 we need to consider what we have learned so far this year.
We need to learnDuring the First Wave of any pandemic we need to over-react, while we find out how the new pathogen behaves and what effects it can have. So, we try everything from social distancing to lock-downs, to see what seems to work in practice. The objective is to reduce the rate of spread of the virus — in biological terms, we are trying to work out what things will flatten the curve (see:
Coronavirus: What is 'flattening the curve,' and will it work?).
For example, one current debate is: do face-masks provide protection, in the community setting? They work in hospitals, for sure (
Face masks really do matter: the scientific evidence is growing), but that is a specialist environment, where they are used by professionals in conjunction with other methods (hand scrubbing, special clothing, etc). We need to find out whether people can routinely wear face-masks properly, so that the masks do what they are designed to do. We may actually be better off with perspex visors, for example, which are also effective at preventing the spread of breath aerosols (which is the main problem), and they can be worn effectively even by a novice — and they do not make us all look like we are involved in a bank hold-up.
We also need different groups of people to try different approaches, to see how effective they are. If everyone does exactly the same thing, strictly following World Health Organization recommendations for example, then we do not learn much, as a global community. That is, a pandemic is simply a widespread (global) series of epidemics, one in each local area. Since countries are all different, culturally, this cultural diversity creates the ideal environment to maximize learning-by-doing, by treating the pandemic as a set of epidemics, to which we might respond differently.
For example, the Buddhist-dominated communities of South-East Asia have done things in a very community-cooperative manner (these people do not work alone, by choice); and they collectively have
the lowest infection rates on the planet. The Muslim-dominated countries of the Middle East do not worry much about life threats (whether they die or not is the Will of Allah), and they collectively have the worst rates. The individual creed of Americans does not encourage them to act co-operatively (resulting in draconian government-mandated lock-downs), and so they also have a very high rate. Sweden is one of the few remaining socialist cultures, where governments give advice rather than issuing instructions (resulting in this case in co-operative self-quarantines), and they have a middling-to-high infection rate.
We learn many things about alternative effective actions from this cultural diversity. In particular, media criticism of the different national reactions to the pandemic is now dying down, as the critics slowly come to realize that uniformity always results in an all-or-none outcome.
What have we learned?Okay, so after the First Wave we know that this new virus can do everything from: apparently nothing (there are plenty of people with antibodies who have never felt any symptoms of having had the virus), to creating flu-like symptoms (
key symptoms: fever, cough, skin rash, loss of taste & smell), on to hospitalization (with usually c. 7 days to get rid of the symptoms but 5 weeks to get rid of the actual virus), or even intensive care (as a result of what is medically called a cytokine storm). For the elderly, and others with pre-existing medical conditions, the virus seems to be one thing too many for their body, the proverbial straw that breaks the camel's back — which can lead to death sooner rather than later.
So, not only does SARS-CoV-2 infection not mean death for the vast majority of people (globally, < 3.6% of reported infections have resulted in death), it does not even necessarily mean sickness at all (eg. a
Swedish study showed that 46% of those study people with antibodies had never reported clinical symptoms). This should mean something for our future responses.
Notably, in those countries where a significant Second Wave is now occurring, the new infections are often not resulting in deaths (except notably in Australia). This is a very important difference between the First and Second Waves, in most places. There is speculation that the SARS-CoV-2 variants currently widespread are less deadly than were those common at the beginning of the pandemic; but it is equally likely that those people who were most susceptible to the virus have already succumbed during the First Wave.
So, we now know about the risk groups, roughly, which is as good as we ever know such things; and we have a good idea about the outcomes of the various risks. This means we can start to do some reasoned things, as a pandemic response. The Second Wave is a perfect time to start treating the Covid-19 situation rationally.
The time for some new action? This means that it is time to start targeting actions to the degree of risk for each person, rather than having over-arching actions that affect everyone equally. Our individual responses to the virus are not equal, so why are most government actions still predicated on the idea that we
are all equal?
The point is, we have to respond to what we have learned about relative risks. For example, I have argued before that the biggest mistake Sweden has made was letting Covid-19 get into the aged-care facilities, which is where most of the country's deaths have now occurred. Has anyone learned from this mistake? Apparently not in the USA:
Untested for Covid-19, nursing-home inspectors move through facilities. Come on people — get your act together.
The response to the First Wave always needs to assume equality, because anything else would be irresponsible, in the face of our initial ignorance. During the Second Wave, however, we are no longer quite so ignorant, and we can tailor our actions to suit the conditions. When are we going to start doing this?
In order to think about this question, it is worthwhile to consider a few topics that seem to be on the agenda, and look at some practical examples of three relevant situations.
Trying to hideAny country that successfully hides from the virus has to keep hiding, forever. New Zealand has recently been crowing about having gone 100 days without a new coronavirus case. That record was destroyed this week (
New Zealand on alert after 4 cases of COVID-19 emerge from unknown source); and it will get even worse on the day they allow the first visitor into their country. Their current Alert Level 3 response cannot change this — you cannot hide from a virus.
New Zealand's near neighbor, Australia, has demonstrated this point even more strongly. In one sense, the Australians understand quarantine, because it is a big part of keeping plant and animal diseases out of their country. For example, international visitors are regularly surprised to have biological products (notably wood) confiscated at the arrival airport — better safe than sorry.
So, dealing with Covid-19 should be straightforward for them — you just apply the same idea to the people, themselves. Sadly, it took them some time to realize that you have to take people straight from the airport to a quarantine hotel, if the quarantine strategy is to work. One of my nephews returned to Sydney (Australia) from Copenhagen (Denmark) at the beginning of the First Wave, and he had to make his own long way by public transport from the airport to the quarantine house that his father had arranged!
So, it should not be a surprise that quarantine has not been effective everywhere in Australia — one mistake is all it takes. This mistake was made in the quarantine hotels in Melbourne (Victoria), where the quarantine security turned out to be a joke (see:
New coronavirus lockdown Melbourne amid sex, lies, quarantine hotel scandal). Perhaps the security guards should have read the earlier article on:
Sex in the time of coronavirus.
The issue here is that Australians are no better than Americans at following government instructions — individual rights take precedence (see:
Individual choice is a bad fit for Covid safety). Even my local newspaper here in Uppsala (Sweden) reported (
Regel brott ger böter) the news that military personnel were sent to visit 3,000 Australians who were supposed to be in self-quarantine at home (due to having tested positive for the virus), and 800 of them (one-quarter!) were not at home. I lived in Australia for 40 years, and this situation surprises me not at all.
So, hiding does not work, long-term, because you have to keep it up for too long to be practical for most people. The Second Wave in Victoria is actually worse than the First Wave, in terms of number of Covid-19 cases. The ensuing lock-down is now even worse than it has been in most other places (see:
'Very dead': army and police patrol the deserted streets of coronavirus-stricken Melbourne); and Victoria itself has been quarantined from the rest of the country.
SchoolsWe have all been told that the effect of Covid-19 is age-related; and the global data shows that this is true everywhere — the older you are, the more likely you are to seriously affected. One outcome of this knowledge is that actions can be tailored to age groups. Notably, we can consider the idea that massively disrupting the lives of very young people may be doing more them harm than good, due to stress if nothing else (
Lockdowns and school shutdowns may make youngsters sicker).
Most countries mandated the closure of schools, and instituted some form of working from home for the pupils. This move was predicated on the idea that children will catch the virus in the crowded schools, and bring the disease home to their elders. This scenario seemed to be the case, for example, in the early spread of the SARS-CoV-2 in northern Italy.
Recent evidence, however, suggests that, while the youngsters do catch the virus, they are much less infectious than older people (see:
COVID-19 study confirms low transmission in educational settings). We are talking about pre-teenagers here, not older children. This does not mean that they can't spread the virus (see:
Latest research points to children carrying, transmitting coronavirus), but merely that this is a much lower risk.
It has therefore been suggested that a rational response would involve a trade-off between disrupting the lives of very young people versus the risk of viral spread (see:
Why it’s (mostly) safe to reopen the schools). Notably, this issue was explicitly considered in Sweden, and during the First Wave it was decided to keep the junior schools open, but to close the senior schools (ie. high school). So, the younger children have all been
trundling off to school every week-day, just as usual, the whole time. As far as I know, there has not been even one reported outbreak involving any of the open schools.
This is why I emphasize the importance of culturally diverse responses to a pandemic. In this case, the Swedes seem to have got it right; and everyone else could learn from this.
Young peopleIt is a different matter for somewhat older (but still young) people. The so-called Millennial generation has had a pretty tough time, especially financially. This is the second financial down-turn that they have experienced in a dozen years, just when they are trying to get themselves onto their own two feet (see:
Millennials slammed by second financial crisis fall even further behind).
So, none of us should be surprised that these people are thoroughly sick of restrictive pandemic responses by now. Indeed, it is becoming widespread news that case rates are increasing among 20-29 year olds (or 15-25, depending on how people are grouped) (see:
WHO urges young people to help control the spread of coronavirus). This has become particularly obvious in Europe (see:
Coronavirus cases rise in Europe as youth hit beaches and bars), but also in North America (see:
B.C. hospitalizations, deaths steady as latest wave hits mostly young people) and Australia (see:
Coronavirus Australia: Why young people are spreading COVID-19).
This is not necessarily as bad as it might sound, because the effect of the virus is age-related, and these people will probably mostly be safe (but not all). The same thing is true for somewhat younger people — youth is a social time, and mandated restrictions about distancing may not be very effective (see:
Why the teenage brain pushes young people to ignore virus restrictions).
Places like Japan and Spain are now cracking down on bars, and the like (eg.
Spain cracks down on outdoor drinking, smoking in renewed push against COVID-19). If you want some survey data on what activities U.S. people currently feel comfortable doing, then check out:
Weekly updates on consumers’ comfort level with various pastimes.
In this situation, Sweden
has not been exempted; and recent coronavirus cases
have become prevalent in the 20-29 year old group, just like elsewhere else. Once again, this emphasizes that our knowledge cannot all come from one place. No-one gets it all right, but they may get some things right; and we should learn from both success
and failure. This is the rational approach, not the one-size-fits-all approach.
Adding to this scenario, as I write this blog post, Europe is having a warm spell (up to 40 °C in the south), and my local newspaper has the headline:
Chaos on Europe's beaches in the heatwave. All governments are warning about the need to continue keeping people apart, for those who wish to avoid infection. Fortunately, the summer holidays are nearing their end in the northern hemisphere.
Concluding commentsFrom the biological perspective, for the future to be bearable, we need to reach herd immunity, which refers to public safety in the presence of a pathogen. This is determined by the proportion of the (local) population that needs to become immunized (either by becoming infected or by being vaccinated) in order for the infection to stop spreading (see:
A new understanding of herd immunity).
We can achieve herd immunity by responding rationally based on the make-up of the population, in terms of the relative risks. At-risk groups need to be protected, while the rest of the people get on with their lives. For example, Stockholm, in Sweden may now
be getting close to herd immunity (or flock immunity, as the locals would call it), the Swedes having foregone the lock-downs imposed elsewhere, and thus allowing immunity to arise naturally.
Herd immunity can be achieved without rationality, of course — we simply wait for the weakest people to die, and the rest are likely to be safe. You might not like the moral implications of doing this, but it is biologically effective, nonetheless. For example, India may potentially end up with the world's worst case-rate for infections, given its population size and large degree of poverty in many areas (where social distancing is not feasible). However, its saving grace, in terms of deaths, may well be the consequent fact that poor people are usually young, because poor people do not live long in the first place. Herd immunity to SARS-CoV-2 is easy to achieve under these circumstances (see:
Herd immunity seems to be developing in Mumbai’s poorest areas).
I vote for the rational approach, myself, among the many biological alternatives.