In a previous post, Guido constructed trees for coronaviruses in the SARS group to search for evidence of recombination. He also constructed unrooted data-display networks using SplitsTree. Here, we discuss our attempts to construct rooted genealogical phylogenetic networks for the same dataset [6] but with some modifications.
In particular, we deleted some sequences, giving a smaller data set with only 12 taxa. These taxa include, next to SARS-CoV-2 (the virus causing COVID-19) and SARS-CoV (responsible for the SARS epidemic in 2002/2003), the viruses MP789 and PCoV_GX-P1E sampled from Malayan pangolins from two different Chinese provinces and several viruses found in different bat species in the horseshoe bat genus (Rhinolophus), all from China.
This research was done by Rosanne Wallin, an MSc student at VU Amsterdam and UvA. Her full thesis as well as all data and results can be found on github.
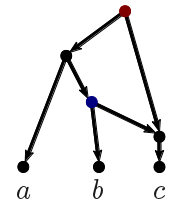
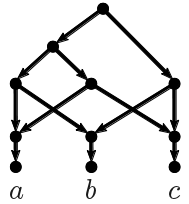
The first algorithm we applied to this data set was the TreeChild Algorithm [1], which is one of the methods that take a number of discordant (rooted, binary) trees as input and finds a rooted network containing each input tree, minimizing the number of reticulate events in the network. To filter out some noise, we contracted some poorly-supported branches and then resolved multifurcations consistently across the trees (using a tool within the TreeChild Algorithm). This gave the network below. Note that the method is restricted to so-called tree-child networks, meaning that certain complex scenarios are excluded (where a network node only has reticulate children). Also note that this is not necessarily the only optimal tree-child network and not all topological differences can be distinguished based on the trees [5].
 |
| Figure 1: Phylogenetic network constructed by the Tree-Child algorithm (blocks_A_len0.01_supp70). |
The network shows no reticulation in the SARS-CoV-2 clade (the bottom four taxa) and puts SARS-CoV-2 right next to RaTG13. Furthermore, it shows a reticulation between an ancestor of HKU3-1 and a common ancestor of SARS-CoV-2 and RaTG13 leading to bat-SL-CoVZC45. However, it cannot exactly identify which common ancestor of SARS-CoV-2 and RaTG13 is the parent, leading to multiple branches (in red) leading into this reticulation. All these observations are consistent with previous research [2].
Importantly, we cannot directly conclude that each reticulation corresponds to a recombination event. See Table 2.1 of David’s book [10] for a nice overview of possible causes of reticulation. Nevertheless, based on [2], it does look like at least the reticulation leading to bat-SL-CoVZC45 corresponds to a recombination event.
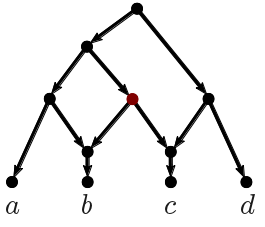
The second algorithm we applied was TriLoNet [3], which constructs a rooted network directly from sequence data. It is restricted to so-called level-1 networks, meaning that it cannot construct overlapping cycles. This method produced the network below.
 |
| Figure 2: Phylogenetic network constructed by TriLoNet. |
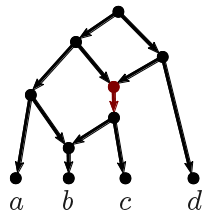
 |
| Figure 3: Phylogenetic network constructed by TriLoNet, after omitting bat-SL-CoVZC45. |