I will be talking at the Festival of Genomics on Wednesday 24 January about Identifying virulence and antimicrobial resistance genes in bacterial using genome-wide association studies. You can preview my talk here.
Posted by: Daniel Wilson
Introducing Doublethink: joint Bayesian-frequentist model-averaged hypothesis testing
This week Nick Arning, Helen Fryer and I released two related preprints describing a new method called Doublethink, and its application to identifying risk factors for COVID-19 hospitalization in UK Biobank:
- Doublethink: simultaneous Bayesian-frequentist model-averaged hypothesis testing. Fryer, Arning, Wilson (2023) arXiv doi: 10.48550/arXiv.2312.17566
- Identifying direct risk factors in UK Biobank with simultaneous Bayesian-frequentist model-averaged hypothesis testing using Doublethink. Arning, Fryer, Wilson (2024) medRxiv doi: 10.1101/2024.01.01.24300687
Doublethink: Bayesian-frequentist model-averaged hypothesis testing
Doublethink enables joint Bayesian and frequentist hypothesis testing when there is model uncertainty by interconverting Bayesian posterior odds and classical (frequentist) p-values. It has broad implications because (i) it reveals connections between the Bayesian approach to model averaging and the classical approach to multiple testing, and (ii) it brings the benefits of Bayesian model averaging to classical statistics.
Doublethink addresses two fundamental problems in hypothesis testing:
- In classical tests, the statistical evidence that one variable directly affects an outcome generally depends on which other variables are assumed to directly affect it.
- In Bayesian tests, the statistical evidence that one variable directly affects an outcome depends on the prior assumptions.
These issues are addressed by computing p-values from Bayesian model-averaged posterior odds, which (1) account for model uncertainty and (2) are theoretically invariant to prior assumptions, assuming large sample sizes.
Doublethink simultaneously controls the frequentist family-wise error rate (FWER) and the Bayesian false discovery rate (FDR). It builds on Johnson's Bayesian tests based on likelihood ratio statistics, and Karamata's theory of regular variation.
Identifying direct risk factors in UK Biobank with Doublethink
We applied Doublethink to identify direct risk factors for COVID-19 hospitalization in UK Biobank. This is a well-studied problem but we took an 'exposome-wide' approach in which we evaluated whether 1,900 variables measured in the UK Biobank each affected the outcome. This is still an under-utilized approach in epidemiology, which usually focuses on candidate risk factors.
Exposome-wide approaches have potential benefits over candidate risk factor approaches, including:
- The ability to discover unexpected results.
- Stringent control for multiple testing.
- Avoidance of bias in choosing candidate risk factors or deciding to publish.
However, we only studied the direct effects of variables on the outcome. This means we cannot make statements about the total (direct and indirect) effects of a variable, e.g. smoking, on the outcome, which are needed in applications like assessing potential interventions.
We identified individual variables and groups of variables that were 'exposome-wide significant' at 9% FDR and 0.05% FWER, after accounting for the direct effects of all other variables.

Comparing our results to over 100 published studies of COVID-19 in UK Biobank, we
- Recapitulated several commonly reported direct risk factors, e.g. age, sex, and obesity.
- Excluded others, e.g. diabetes, cardiovascular disease, and hypertension, which might be mediated through other variables that measure general comorbidity.
- Identified some infrequently reported direct risk factors, both individually, e.g. lung infection, and as groups, e.g. constipation/urinary tract infection, which might reflect underlying kidney disease.
The ability to test groups of variables, which increases sensitivity, was one of the benefits of Doublethink's model-averaging approach. It is particularly helpful in large biobanks that measure thousands of variables, because correlation between variables is pervasive, and can dilute the significance of individual variables that measure similar phenomena, like the numerous types of deprivation index. It serves as a flexible alternative to pre-analysis variable filtering algorithms, while controlling the risk of false positives by pre-defining significance thresholds for all possible tests.
Posted by in covid 19, Doublethink, FDR, FWER, Helen Fryer, Nicolas Arning, UK Biobank
Rewley House Lecture: Role of data science in the pandemic
This year I was invited to give the Rewley House Lecture, a multidisciplinary research talk open to all, at the Department for Continuing Education, where I am Director of Studies in Data Science.
I talked about how data science has been used during the COVID-19 pandemic, spanning vaccine design, clinical trials, surveillance and policy advice, and highlighting the identification of risk factors for disease.
If you like this talk, you might be interested in the following courses available this academic year:
- Infectious Disease Modelling: Mathematical Techniques (September 2022)
- Infectious Disease Modelling: Applied Methods in R (January 2023)
- Pandemic Data Science (April 2023)
Posted by in Continuing Education, covid 19, talk
Identifying resistance genes in tuberculosis
Newly published in PLOS Biology is our work identifying genes that confer resistance to common and last-resort antibiotics in bacteria that cause tuberculosis. Resistance to these drugs contributes to mortality and sickness on a pandemic scale every year, and disproportionately affects the poorest people in the world.
This new article is one of a series presenting results generated by more than 100 scientists across 23 countries across 5+ years as part of a collaboration called CRyPTIC.
Our role in CRyPTIC was the discovery of genes and mutations likely to cause drug resistance by applying a tool known as a genome-wide association study (GWAS), an approach we helped adapt to bacteria.
Using GWAS, we identified previously uncatalogued genes and mutations underlying resistance to every one of the 13 drugs we investigated. These include new and repurposed drugs, as well as the first- and second-line drugs more often used to treat tuberculosis.
Thanks to its generous funders, CRyPTIC dedicated scale (10,000+ genomes) and technical innovation (new high-throughput MIC assays) to help decode the DNA blueprint of antibiotic resistance. Pushing these boundaries has yielded a steep increase of up to 36% in the variation in resistance attributable to the genome for the important and previously understudied new and repurposed drugs.
Science at this scale can produce a seemingly overwhelming wealth of new information. We avoided the temptation to over-emphasize any individual result for the sake of simple narrative. Instead, we highlighted discoveries of uncatalogued genes or genetic variants that we found for every drug investigated:
• The amidase AmiA2 and GTPase Era for bedaquiline.
• The cytochrome P450 enzyme Cyp142 for clofazimine.
• The serine/threonine protein kinase PknH for delaminid.
• The antitoxin VapB20 for linezolid.
• The PPE-motif family outer membrane protein PPE42 for amikacin and kanamycin.
• The antibiotic-induced transcriptional regulator WhiB7 for ethionamide.
• The rRNA methylase TlyA for levofloxacin.
• The DNA gyrase subunit B GyrB for moxifloxacin.
• The putative rhodaneses CysA2 and CysA3 for rifabutin.
• The tRNA/rRNA methylase SpoU for ethambutol and rifampicin.
• The multidrug efflux transport system repressor Rv1219 for isoniazid.
All these hits passed stringent evidence thresholds that take into account the large amount of data crunched. For each hit, we identified possible relationships between gene functions, such as they are known, and the mechanism of action of the antibiotics.
Beyond the biological discoveries of primary interest, this new paper unveils methodological advances in bacterial GWAS. We introduced a systematic, whole-genome approach to analysing not just short DNA sequences (so called oligonucleotide or “kmer”-based approaches), but also short sequences of the proteins that the DNA codes for (an oligopeptide-based approach). We have released our software on an open-source GitHub repository.
We also discovered a relationship that may help disentangle a technical issue in bacterial GWAS where the co-occurence of traits can trick us into thinking that a gene influences one trait when it influences another instead. For antimicrobial resistance, this issue is known as artefactual cross resistance. We observed that true associations tended to produce larger associations (as measured by the 'coefficient', rather than the p-value), providing a possible way to prioritize signals in the future.
This paper was published alongside the CRyPTIC Data Compendium in PLOS Biology, in which we released our data open source to the community, with resources provided by the European Bioinformatics Institute.
Some of the results of CRyPTIC have already been rushed into service by the World Health Organization on the grounds of exceptional importance based on a candidate gene approach; this includes the DNA gyrase subunit B – moxifloxacin association spotlighted above (Walker et al 2022). However, the new results go beyond a candidate gene approach, detecting a range of previously uncatalogued genes via its agnostic, whole-genome strategy.
Unpicking the genetics of antimicrobial resistance is a priority for improving rapid susceptibility tests for individual patients, selecting drug regimens that inhibit the evolution of multidrug resistance, and developing improved treatment options. The need is particularly great in M. tuberculosis, which killed 1.4 million people in 2019, owing to the slow (6-12 week) turnaround of traditional susceptibility testing, and the alarming threat of multidrug resistant tuberculosis. The discovery of many new candidate resistance variants therefore represents an advance that we hope will contribute to progress in reducing the burden of disease.
Seeking Postdoc in Statistical Genetics and Infectious Disease
I am seeking a senior postdoc in Statistical Genetics and Infectious Disease to join my research group at the Big Data Institute, University of Oxford. Our research into Infectious Disease Genomics is focused on developing and applying big data methods to identify genetic risk factors for disease, both microbial virulence factors and human susceptibility genes. We are focused on a range of bacterial and viral diseases including staphylococcal sepsis and COVID-19.
The Big Data Institute, part of Oxford Population Health, provides an excellent environment for multi-disciplinary research and teaching. Situated on the modern Old Road Campus in the heart of the medical sciences neighbourhood of Headington, we benefit from outstanding facilities and opportunities to collaborate with world-leading scientists and clinicians to help expand knowledge and improve global health.
As a Senior Postdoc the post-holder will work closely with me to jointly lead the implementation, design and application of new statistical tools for genome-wide association studies, and to lead the biological interpretation of key findings. They will develop novel methodologies for analysis and data collection, take the lead in the production of scientific reports and publications and supervise junior group members.
To be considered applicants will have a PhD and post-doctoral experience in a relevant subject, with direct experience in statistical genetics, demonstrable expertise and knowledge of the statistical genetics literature or a closely related, relevant discipline and a publication record as first author, in statistical genetics.
The position is full time (part time considered) and fixed-term for 3 years.
The closing date for application is 12.00 noon GMT on 18th March.
Click here for more information including how to apply.
Posted by in Big Data Institute, Genomics, infection, Postdoc, Recruitment, statistics
Announcing the Oxford Statistical Genomics Summer School 2022
Join us at St Hilda's College Oxford, overlooking the River Cherwell and Christ Church Meadow, for an immersive week-long residential post-graduate summer school on Statistical Genomics on 19th-24th June 2022. This course aims to connect post-graduate and post-doctoral researchers from academia and industry with experts at Oxford's Big Data Institute, Wellcome Centre for Human Genetics, and Department of Statistics.
Our friendly tutors, internationally recognised for their scientific expertise, will offer specialist instruction and hands-on computer practicals across five broad areas of Statistical Genomics: Next-generation Sequence Data Analysis, Gene and Variant Association Testing, Genomics of Infectious Diseases, Genealogical Inference and Analysis, and Medical Genomics.
The course is aimed at trainee scientists actively engaged in statistical genomics research, who wish to expand their knowledge of concepts and techniques.
Click here for more information including how to apply.

Posted by in Statistical Genomics Summer School, teaching
Postdoctoral and Ph.D. positions in the group
If you are interested in joining the group, please contact me (details here) with a brief explanation and a copy of an up-to-date CV.
Posted by in Uncategorized
Announcing ProbGen22 in Oxford 28-30 March
The organizing committee is pleased to announce the 7th Probabilistic Modeling in Genomics Conference (ProbGen22) to be held at the Blavatnik School of Government and Somerville College Oxford from 28th-30th March 2022.
The meeting will be a hybrid in-person and online event. Talk sessions will feature live speakers, both in-person and online, and will take place during the afternoons (making live attendance feasible for US timezones). Talks will be recorded and made available to registrants for a period of one month. Poster sessions will be held online during the evenings.
The conference will cover probabilistic models, algorithms, and statistical methods across a broad range of applications in genetics and genomics. We invite abstract submissions on a range of topics including population genetics, natural selection, Quantitative genetics, Methods for GWAS, Applications to cancer and other diseases, Causal inference in genetic studies, Functional genomics, Assembly and variant identification, Phylogenetics, Single cell 'omics, Deep learning in genomics and Pathogen genomics.
The registration deadline is 28th February 2022.
For more details visit the conference website.
Posted by in conference, GWAS, Population Genetics, ProbGen, Selection, statistics
Two new positions: Senior Statistical Geneticist and Bioinformatician
Two new positions are available in my Infectious Disease Genomics group at the Big Data Institute, University of Oxford.
A Senior Postdoctoral Statistical Geneticist to jointly lead the implementation, design and application of new statistical tools for genome-wide association studies, lead the biological interpretation of key findings, develop methodologies and supervise junior group members. This post would suit a candidate with a PhD and relevant post-doctoral experience including direct experience in statistical genetics. Candidates without post-doctoral experience may be considered for a less senior appointment.
A Bioinformatician to provide expertise for computationally intensive analyses including genome-wide association studies and RNAseq studies of differential gene expression, as well as contributing to informatics projects as part of a wider collaboration with national biomedical cohorts. This post would suit a candidate with either a post-graduate degree related to Bioinformatics, Statistics, and Computing or equivalent experience in industry.
The application deadline for both posts is Noon GMT on Friday 7th January 2022.
Posted by in Big Data Institute, Bioinformatics, GWAS, Postdoc, Recruitment, statistics
New paper: Machine learning to predict the source of campylobacteriosis using whole genome data
This study, published in October in PLOS Genetics, brings together machine learning, large bacterial isolate collections and whole genome sequencing to address the general problem of how to trace the source of human infections.
Specifically, we investigated campylobacteriosis, a common infection of animal origin causing ~1.5 million cases of gastroenteritis and 10,000 hospitalizations every year in the United States alone. We show that our combined machine learning/genomics analyses:
- Improve the accuracy with which infections can be traced back to farm reservoirs.
- Identify evolutionary shifts in bacterial affinity for livestock host species.
- Detect changes in human infection capability within related strains.
These results will improve understanding not only of Campylobacter, but more generally as these technologies can readily be applied to other important bacterial pathogen species.
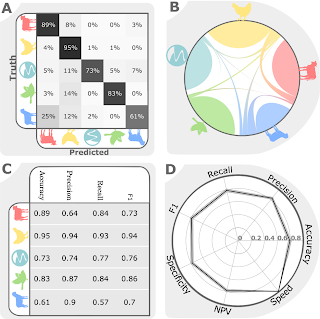
This paper builds on previous work published by the group, including our well cited Tracing the source of campylobacteriosis (Wilson et al 2008, PLOS Genetics 4:e1000203). The use of these methods for tracing infection has influenced public health policy and contributed to reducing disease burden.
This work demonstrates the potential for modern genomics and artificial intelligence approaches to address common and serious problems that affect our everyday lives. The awareness of the importance of infection to society has rarely been higher than in 2021, and while the current pandemic imposes an acute global problem, other infections continue to present long-term threats to health and productivity.